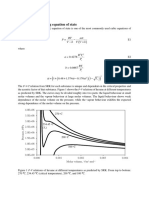
You might also like
- r05321802 Non Ferrous Extractive MetallurgyDocument6 pagesr05321802 Non Ferrous Extractive MetallurgySRINIVASA RAO GANTANo ratings yet
- Structure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsFrom EverandStructure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsNo ratings yet
- Metallurgical Physical ChemistryDocument45 pagesMetallurgical Physical ChemistryAlvin Garcia PalancaNo ratings yet
- Butts A. Metallurgical Problems (027-032)Document6 pagesButts A. Metallurgical Problems (027-032)Miguel Lopez BarretoNo ratings yet
- ch4-6 SlipDocument5 pagesch4-6 SlipAyesha Farooq100% (1)
- Nucleation and Growth of Metals: From Thin Films to NanoparticlesFrom EverandNucleation and Growth of Metals: From Thin Films to NanoparticlesNo ratings yet
- Metals: Hydrogen Ironmaking: How It WorksDocument15 pagesMetals: Hydrogen Ironmaking: How It WorksMarko's Brazon'No ratings yet
- 3 - Metallurgical Thermodynamics and KineticsDocument120 pages3 - Metallurgical Thermodynamics and KineticsMollin Siwella100% (1)
- Transition Metal ToxicityFrom EverandTransition Metal ToxicityG. W. RichterNo ratings yet
- 2-Conduction in SolidDocument46 pages2-Conduction in SolidRichard Adventus HutasoitNo ratings yet
- Metallurgical Volume 1Document196 pagesMetallurgical Volume 1Ugadi SunilNo ratings yet
- Synthesis and Fabrication of AlloysDocument61 pagesSynthesis and Fabrication of AlloysSURAJ NAGNo ratings yet
- Solidification of Metals (To Be Completed) : Prof. H. K. Khaira Professor, Deptt. of MSME M.A.N.I.T., BhopalDocument62 pagesSolidification of Metals (To Be Completed) : Prof. H. K. Khaira Professor, Deptt. of MSME M.A.N.I.T., BhopalIndranil Bhattacharyya100% (1)
- Deformation & Strengthening Mechanisms: Issues To Address..Document48 pagesDeformation & Strengthening Mechanisms: Issues To Address..Christopher MurilloNo ratings yet
- 20230116-MT-205-PD-BNS-L-4 To L-6 (2022-2023) Notes - 2Document47 pages20230116-MT-205-PD-BNS-L-4 To L-6 (2022-2023) Notes - 2Kaustav SaikiaNo ratings yet
- TernaryDocument8 pagesTernaryNino Rico LatupanNo ratings yet
- Equilibrium Crystallisation of Iron-Carbon Alloys LectureDocument25 pagesEquilibrium Crystallisation of Iron-Carbon Alloys LectureTisza_MNo ratings yet
- Advances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenFrom EverandAdvances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenW. HoppeNo ratings yet
- Floatation of Sulphide OresDocument49 pagesFloatation of Sulphide OresRavian Lhr100% (1)
- Principles of Extractive MetallurgyDocument128 pagesPrinciples of Extractive MetallurgyFelix TinasheNo ratings yet
- Ternary Phase DiagramsDocument40 pagesTernary Phase Diagramsviky kava100% (1)
- Janusz S Laskowski PDFDocument178 pagesJanusz S Laskowski PDFjoseph ayronNo ratings yet
- Current Topics in Amorphous Materials: Physics & TechnologyFrom EverandCurrent Topics in Amorphous Materials: Physics & TechnologyY. SakuraiRating: 5 out of 5 stars5/5 (1)
- Physical Electronics Slides of Chapter 7 All SlidesDocument45 pagesPhysical Electronics Slides of Chapter 7 All SlidesLina Al-SalehNo ratings yet
- Gate 2013 Metallurgical Engineering SolutionDocument10 pagesGate 2013 Metallurgical Engineering SolutionAMMASI A SHARAN100% (7)
- The ACORGA® OPT Series: Comparative Studies Against Aldoxime: Ketoxime ReagentsDocument10 pagesThe ACORGA® OPT Series: Comparative Studies Against Aldoxime: Ketoxime ReagentsVictor Alberto Solano GaviñoNo ratings yet
- The Rewards of Patience, Scheffel 2006Document9 pagesThe Rewards of Patience, Scheffel 2006NatitoPazGonzalezArmijoNo ratings yet
- A Coupled Fluid Dynamic-Discrete Element Simulation of Heat and Mass Transfer in A Lime Shaft KilnDocument14 pagesA Coupled Fluid Dynamic-Discrete Element Simulation of Heat and Mass Transfer in A Lime Shaft KilnGuglielmo CancelliNo ratings yet
- Analytical Chemistry of Zirconium and Hafnium: International Series of Monographs in Analytical ChemistryFrom EverandAnalytical Chemistry of Zirconium and Hafnium: International Series of Monographs in Analytical ChemistryNo ratings yet
- Engineering Measurements - Methods and Intrinsic Errors - WILLEY PDFDocument195 pagesEngineering Measurements - Methods and Intrinsic Errors - WILLEY PDFSuci SetianingsihNo ratings yet
- Microstructural Development - The Science and Design of Engineering MaterialsDocument62 pagesMicrostructural Development - The Science and Design of Engineering MaterialsPurin Suttasit100% (1)
- Amorphous and Nano Alloys Electroless Depositions: Technology, Composition, Structure and TheoryFrom EverandAmorphous and Nano Alloys Electroless Depositions: Technology, Composition, Structure and TheoryNo ratings yet
- Resistance and Deformation of Solid Media: Pergamon Unified Engineering SeriesFrom EverandResistance and Deformation of Solid Media: Pergamon Unified Engineering SeriesNo ratings yet
- Preparative Methods in Solid State ChemistryFrom EverandPreparative Methods in Solid State ChemistryPaul HagenmullerNo ratings yet
- Physical Metallurgy M1 PDFDocument21 pagesPhysical Metallurgy M1 PDFAnca ElenaNo ratings yet
- Dislocation Mobility and Hydrogen - A Brief ReviewDocument6 pagesDislocation Mobility and Hydrogen - A Brief ReviewIchsan HakimNo ratings yet
- Solidification of Single-Phase Alloys - 2007Document12 pagesSolidification of Single-Phase Alloys - 2007pkn_pnt9950No ratings yet
- ECE 3040 Lecture 22: Numerical Solution of Differential EquationsDocument34 pagesECE 3040 Lecture 22: Numerical Solution of Differential EquationsAqe KitamNo ratings yet
- Surface Physics of Materials: Materials Science and TechnologyFrom EverandSurface Physics of Materials: Materials Science and TechnologyNo ratings yet
- Ch09 Diagramas de FasesDocument38 pagesCh09 Diagramas de FasesDennys Quimi BorborNo ratings yet
- Electronic Absorption Spectra and Geometry of Organic Molecules: An Application of Molecular Orbital TheoryFrom EverandElectronic Absorption Spectra and Geometry of Organic Molecules: An Application of Molecular Orbital TheoryRating: 5 out of 5 stars5/5 (1)
- Principles of Metallurgical OperationsDocument38 pagesPrinciples of Metallurgical OperationsMuhammad Saim100% (1)
- Semiconductor Manufacturing TechnologyDocument38 pagesSemiconductor Manufacturing Technologymustopa Dwi Ramadhany Semester 3No ratings yet
- Extraction and Selective Purification of Gallium (III), Vanadium (IV) From Aluminum (III) Contained Acid Sulphate Solutions Using D2ehfaDocument5 pagesExtraction and Selective Purification of Gallium (III), Vanadium (IV) From Aluminum (III) Contained Acid Sulphate Solutions Using D2ehfaInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- The Physical Metallurgy of Fracture: Fourth International Conference on Fracture, June 1977, University of Waterloo, CanadaFrom EverandThe Physical Metallurgy of Fracture: Fourth International Conference on Fracture, June 1977, University of Waterloo, CanadaD M R TaplinNo ratings yet
- Materials Principles and Practice: Electronic Materials Manufacturing with Materials Structural MaterialsFrom EverandMaterials Principles and Practice: Electronic Materials Manufacturing with Materials Structural MaterialsCharles NeweyNo ratings yet
- Midterm Review SolutionsDocument16 pagesMidterm Review SolutionsKate SongNo ratings yet
- Structural Analysis of Hexaferrite Materials - Yazan MaswadehDocument112 pagesStructural Analysis of Hexaferrite Materials - Yazan MaswadehYazan MaswadehNo ratings yet
- Lab Safety Design ManualDocument4 pagesLab Safety Design ManualHariom GuptaNo ratings yet
- P6 Training Manual Encore Group PDFDocument142 pagesP6 Training Manual Encore Group PDFPd Proxy100% (1)
- Brochure - Gas Power PlantsDocument16 pagesBrochure - Gas Power PlantsGonzalo Bevilacqua AlenNo ratings yet
- U D C 3 5 0 0 A P P L I C A T I o N N o T eDocument4 pagesU D C 3 5 0 0 A P P L I C A T I o N N o T earjun232No ratings yet
- Mark and EllenDocument45 pagesMark and EllenHariom GuptaNo ratings yet
- Motion-Man and Object Changes Its Psition Continously WithDocument2 pagesMotion-Man and Object Changes Its Psition Continously WithHariom GuptaNo ratings yet
- Lab Safety Design ManualDocument4 pagesLab Safety Design ManualHariom GuptaNo ratings yet
- Approved vendor list requestDocument10 pagesApproved vendor list requestHariom GuptaNo ratings yet
- 225Mw Combined Cycle Gas Based Power Pla: Files Exported On 11/19/2012Document18 pages225Mw Combined Cycle Gas Based Power Pla: Files Exported On 11/19/2012Hariom GuptaNo ratings yet
- Boiler Drum Level Measurement and ControlDocument13 pagesBoiler Drum Level Measurement and ControlasdmoomNo ratings yet
- Object XMLDocumentDocument114 pagesObject XMLDocumentprabhuleanNo ratings yet
- TechniquesDocument29 pagesTechniquesHariom Gupta100% (1)
- Ch4 - Gas CuttingDocument26 pagesCh4 - Gas CuttingkevinjunNo ratings yet
- Air Cooled CondenserDocument20 pagesAir Cooled CondenserVivek SinghNo ratings yet
- Ideal Gas Vs Real GasDocument9 pagesIdeal Gas Vs Real GasRaymond BaldelovarNo ratings yet
- Principles of Thermodynamics NotesDocument12 pagesPrinciples of Thermodynamics NotesSaniya SohailNo ratings yet
- SRK Equation of State ExplainedDocument5 pagesSRK Equation of State Explainednutnicha suwannangkoonNo ratings yet
- Physical Chemistryii PHC115B: DR Nthabiseng Ntholeng 2020Document43 pagesPhysical Chemistryii PHC115B: DR Nthabiseng Ntholeng 2020Enabewhkom OhpmNo ratings yet
- Calculate Density Using Ideal Gas LawDocument12 pagesCalculate Density Using Ideal Gas LawJasminSutkovicNo ratings yet
- Properties of Pure SubstanceDocument54 pagesProperties of Pure SubstancesitinurhanizaNo ratings yet
- Physics GRE SolutionsDocument338 pagesPhysics GRE SolutionsDavid Latchman100% (2)
- Real Gas Flow Simulation in Damaged Distribution PipelinesDocument8 pagesReal Gas Flow Simulation in Damaged Distribution PipelinesJorge Andrés Palacio CardonaNo ratings yet
- Lecture 4 Gas Laws and RelationsDocument28 pagesLecture 4 Gas Laws and RelationsArsal SohrabNo ratings yet
- William C. Reynolds, Piero Colonna - Thermodynamics - Fundamentals and Engineering Applications-Cambridge University Press (2018)Document421 pagesWilliam C. Reynolds, Piero Colonna - Thermodynamics - Fundamentals and Engineering Applications-Cambridge University Press (2018)Vipul JoshiNo ratings yet
- ENSC 461 Tutorial, Week#4 - IC EnginesDocument8 pagesENSC 461 Tutorial, Week#4 - IC Enginesandres179No ratings yet
- CP de VaporDocument6 pagesCP de VaporNatalia MorenoNo ratings yet
- Gaseous State and ThermodynamicsDocument45 pagesGaseous State and ThermodynamicsJhon HarrisonNo ratings yet
- HEAT LAWSDocument29 pagesHEAT LAWSMohib UddinNo ratings yet
- Gases: 5/75 Questions in Multiple Choice Almost Every Year in Free Response SectionDocument53 pagesGases: 5/75 Questions in Multiple Choice Almost Every Year in Free Response SectionZenobia Joy VillarbaNo ratings yet
- Joule-Thomson Coefficient of Carbon DioxideDocument5 pagesJoule-Thomson Coefficient of Carbon DioxideRuth UmerezNo ratings yet
- MIN 106: ENGINEERING THERMODYNAMICS Tutorial Sheet 3 Properties of Pure SubstancesDocument2 pagesMIN 106: ENGINEERING THERMODYNAMICS Tutorial Sheet 3 Properties of Pure Substancestanisha guptaNo ratings yet
- Dummy 331Document2 pagesDummy 331MOHAMMA MUSANo ratings yet
- Batang E, GheremyDocument3 pagesBatang E, Gheremyyeng botzNo ratings yet
- BEC198-1 PrelimsDocument98 pagesBEC198-1 PrelimsClash Nikov100% (1)
- Ideal Gas ProcessesDocument71 pagesIdeal Gas ProcessesAnand NadgireNo ratings yet
- Chem Academy: ThermodynamicsDocument5 pagesChem Academy: ThermodynamicsHamit RanaNo ratings yet
- GAS LAWS AND THERMODYNAMICS EQUATIONSDocument5 pagesGAS LAWS AND THERMODYNAMICS EQUATIONSSyed Mairaj Ul HaqNo ratings yet
- A Level Chap 5 EOCDocument4 pagesA Level Chap 5 EOCTrúc Hồ100% (1)
- Thermodynamics For Beginners - Chapter 5 WORKING WITH IDEAL GASDocument35 pagesThermodynamics For Beginners - Chapter 5 WORKING WITH IDEAL GASDudu MamanNo ratings yet
- BE 8256 - Basic Mechanical Engineering / U – II / Compiled by R.Arul KamalakumarDocument24 pagesBE 8256 - Basic Mechanical Engineering / U – II / Compiled by R.Arul KamalakumararulrakkNo ratings yet
- CHE 133 Lab #2Document4 pagesCHE 133 Lab #2Ben Killam100% (1)
- University of Illinois at Chicago Department of Physics: Thermodynamics & Statistical Mechanics Qualifying ExaminationDocument5 pagesUniversity of Illinois at Chicago Department of Physics: Thermodynamics & Statistical Mechanics Qualifying ExaminationrujintoNo ratings yet
- UPCS Physics Problem SetDocument13 pagesUPCS Physics Problem SetRicardo Jose BracamonteNo ratings yet
- Background: EM/ASSIGNMENT (1) 2017Document10 pagesBackground: EM/ASSIGNMENT (1) 2017neenoonaaNo ratings yet