You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Introduction To Molecular Orbital TheoryDocument19 pagesIntroduction To Molecular Orbital TheoryChinni YalamanchiliNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Book Start PDF PagesDocument509 pagesBook Start PDF PagesJoaquín SantiagoNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- Structures of Simple Binary CompoundsDocument16 pagesStructures of Simple Binary CompoundsJoaquín SantiagoNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Mechanistic Basis For High Reactivity of (Salen) Co-OTs in The Hydrolytic Kinetic Resolution of Terminal EpoxidesDocument10 pagesMechanistic Basis For High Reactivity of (Salen) Co-OTs in The Hydrolytic Kinetic Resolution of Terminal EpoxidesJoaquín SantiagoNo ratings yet
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Inorganic ChemistryDocument7 pagesInorganic ChemistryJoaquín SantiagoNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Glycerol As An Alternative Green Medium For Carbonyl Compound ReductionsDocument8 pagesGlycerol As An Alternative Green Medium For Carbonyl Compound ReductionsJoaquín SantiagoNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Liber Les 1977Document4 pagesLiber Les 1977Joaquín SantiagoNo ratings yet
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Cu Nico 1982Document3 pagesCu Nico 1982Joaquín SantiagoNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Advantages - Microwave SynthesisDocument2 pagesAdvantages - Microwave SynthesisJoaquín SantiagoNo ratings yet
- Fundamentals of Ion ExchangeDocument9 pagesFundamentals of Ion Exchangewaheed2286No ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Polycyclic and Heterocyclic Aromatic CompoundsDocument46 pagesPolycyclic and Heterocyclic Aromatic CompoundsAhmed Mohamed IbrahimNo ratings yet
- DNSDHR 3 y 4 T 34 y 475 B 4Document6 pagesDNSDHR 3 y 4 T 34 y 475 B 4Joaquín SantiagoNo ratings yet
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- IntroductionDocument16 pagesIntroductionJoaquín SantiagoNo ratings yet
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Photoreduction of Benzophenones by Amines in Room-Temperature Ionic LiquidsDocument3 pagesPhotoreduction of Benzophenones by Amines in Room-Temperature Ionic LiquidsJoaquín SantiagoNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- A New Synthesis of Aldehydes - Henry StephenDocument4 pagesA New Synthesis of Aldehydes - Henry StephenJoaquín SantiagoNo ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Tetrazole - Wikipedia, The Free EncyclopediaDocument3 pagesTetrazole - Wikipedia, The Free EncyclopediaJoaquín SantiagoNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- DhbfadhfbdjasbjsbdfDocument12 pagesDhbfadhfbdjasbjsbdfJoaquín SantiagoNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- GX 6 DbsxfydfsyuatdfubdtfDocument9 pagesGX 6 DbsxfydfsyuatdfubdtfJoaquín SantiagoNo ratings yet
- Shdgwqytewu ShdgwqytewuDocument3 pagesShdgwqytewu ShdgwqytewuJoaquín SantiagoNo ratings yet
- SgahsDocument16 pagesSgahsJoaquín SantiagoNo ratings yet
- Water PDFDocument3 pagesWater PDFJoaquín SantiagoNo ratings yet
- Ajsdhsdgwiyer 8 Ew 02348375Document6 pagesAjsdhsdgwiyer 8 Ew 02348375Joaquín SantiagoNo ratings yet
- XV Olimpiada Nacional de Química MX SolucionesDocument10 pagesXV Olimpiada Nacional de Química MX SolucionesJoaquín SantiagoNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Hola HolaDocument51 pagesHola HolaJoaquín SantiagoNo ratings yet
- Qwertyuiop QwertyuiopDocument5 pagesQwertyuiop QwertyuiopJoaquín SantiagoNo ratings yet
- Qwertyuiop QwertyuiopDocument5 pagesQwertyuiop QwertyuiopJoaquín SantiagoNo ratings yet
- Qwertyuiop QwertyuiopDocument5 pagesQwertyuiop QwertyuiopJoaquín SantiagoNo ratings yet
- HOLA22Document14 pagesHOLA22Joaquín SantiagoNo ratings yet
- HOLA22Document14 pagesHOLA22Joaquín SantiagoNo ratings yet
- Dual Nature of LightDocument15 pagesDual Nature of LightUriahs Victor75% (4)
- Influence of Dirt Accumulation On Performance of PV Panels: SciencedirectDocument7 pagesInfluence of Dirt Accumulation On Performance of PV Panels: SciencedirectEsra AbdulhaleemNo ratings yet
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Measuring Instruments Practice QuestionsDocument6 pagesMeasuring Instruments Practice QuestionsAsif Ayaz100% (2)
- Equilibrium Stage Processes - Docx CetDocument25 pagesEquilibrium Stage Processes - Docx CetPortia ShilengeNo ratings yet
- Mathematical Methods in Engineering and ScienceDocument1,474 pagesMathematical Methods in Engineering and ScienceSoupramanien KathirvelouNo ratings yet
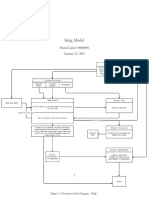
- Ising ModelDocument5 pagesIsing Modeldecerto252No ratings yet
- HW1Document2 pagesHW1Jimmy LauNo ratings yet
- Fortran (For NACA)Document7 pagesFortran (For NACA)hdslmnNo ratings yet
- Experiment #2: Continuous-Time Signal Representation I. ObjectivesDocument14 pagesExperiment #2: Continuous-Time Signal Representation I. ObjectivesMarvin AtienzaNo ratings yet
- CPM cc1 - CH 2 VocabularyDocument3 pagesCPM cc1 - CH 2 Vocabularyapi-252110147No ratings yet
- Column Design PDFDocument2 pagesColumn Design PDFRobin0% (1)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Simulation of Soil Compaction With Vibratory Rollers PDFDocument13 pagesSimulation of Soil Compaction With Vibratory Rollers PDFSandeep KumarNo ratings yet
- Lahore University of Management Sciences: EE539 - Radar SystemsDocument3 pagesLahore University of Management Sciences: EE539 - Radar SystemsDr-Raghad Al-FahamNo ratings yet
- Liquid Liquid SeparatorsDocument26 pagesLiquid Liquid SeparatorsAjay Pratap SinghNo ratings yet
- High-Efficiency Solar Inverter Test Report SummaryDocument17 pagesHigh-Efficiency Solar Inverter Test Report Summarybharath prabhuNo ratings yet
- Molecular Partition Function ExplainedDocument18 pagesMolecular Partition Function ExplainedAnimasahun Olamide HammedNo ratings yet
- Lecture 1Document33 pagesLecture 1fatimaasiriNo ratings yet
- ASTM TablesDocument185 pagesASTM Tableslahiru198367% (3)
- Jacobs Co Algebra IntroDocument190 pagesJacobs Co Algebra IntrozmthNo ratings yet
- Module - 6 - Slides - Part 1 PDFDocument124 pagesModule - 6 - Slides - Part 1 PDFvarniktpNo ratings yet
- Normal Distribution Giuded SolutionsDocument5 pagesNormal Distribution Giuded SolutionsleeshanghaoNo ratings yet
- Properties of CompoundsDocument15 pagesProperties of CompoundsPrasad YarraNo ratings yet
- Archimedes Heat RayDocument5 pagesArchimedes Heat RayElliah Jen BiluganNo ratings yet
- C680Document22 pagesC680dinhtung2210100% (2)
- Microplan - Katalog 2016 ENDocument55 pagesMicroplan - Katalog 2016 END.T.No ratings yet
- Alloy ZN Ni PDFDocument8 pagesAlloy ZN Ni PDFElenaNo ratings yet
- 16.323 Principles of Optimal Control: Mit OpencoursewareDocument26 pages16.323 Principles of Optimal Control: Mit Opencoursewaremousa bagherpourjahromiNo ratings yet
- SA Engineering College Statistics and Numerical Methods NotesDocument8 pagesSA Engineering College Statistics and Numerical Methods NotesRaja Nirmal KumarNo ratings yet
- Adaptive Followup Mastering PhysicsDocument8 pagesAdaptive Followup Mastering PhysicsElloani Ross Arcenal PitogoNo ratings yet
- Material Balance On A 2 Unit DistillationsDocument6 pagesMaterial Balance On A 2 Unit Distillationsnhalieza1067No ratings yet
- Quantum Physics: What Everyone Needs to KnowFrom EverandQuantum Physics: What Everyone Needs to KnowRating: 4.5 out of 5 stars4.5/5 (48)
- Quantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessFrom EverandQuantum Spirituality: Science, Gnostic Mysticism, and Connecting with Source ConsciousnessRating: 4 out of 5 stars4/5 (6)
- Quantum Physics for Beginners Who Flunked Math And Science: Quantum Mechanics And Physics Made Easy Guide In Plain Simple EnglishFrom EverandQuantum Physics for Beginners Who Flunked Math And Science: Quantum Mechanics And Physics Made Easy Guide In Plain Simple EnglishRating: 4.5 out of 5 stars4.5/5 (18)
- Too Big for a Single Mind: How the Greatest Generation of Physicists Uncovered the Quantum WorldFrom EverandToo Big for a Single Mind: How the Greatest Generation of Physicists Uncovered the Quantum WorldRating: 4.5 out of 5 stars4.5/5 (8)
- Summary and Interpretation of Reality TransurfingFrom EverandSummary and Interpretation of Reality TransurfingRating: 5 out of 5 stars5/5 (5)
- The Tao of Physics: An Exploration of the Parallels between Modern Physics and Eastern MysticismFrom EverandThe Tao of Physics: An Exploration of the Parallels between Modern Physics and Eastern MysticismRating: 4 out of 5 stars4/5 (500)