You might also like
- Presentation 1Document60 pagesPresentation 1Mezgebu BiresawNo ratings yet
- Química Inorgánica II - Parte 4 - Tipos de Enlace y Reactividad - 21PDocument91 pagesQuímica Inorgánica II - Parte 4 - Tipos de Enlace y Reactividad - 21Pandrea davila alvarezNo ratings yet
- Sec-A Part 5Document3 pagesSec-A Part 5Bhavesh GargNo ratings yet
- Crystal Field TheoryDocument58 pagesCrystal Field TheoryAakash GuptaNo ratings yet
- TextoDocument13 pagesTextoFRANSNo ratings yet
- I PUC Model QP AnswerDocument8 pagesI PUC Model QP AnswerSamanth PattarNo ratings yet
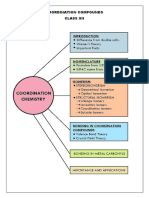
- Introduction To Coordination ChemistryDocument15 pagesIntroduction To Coordination ChemistryDnyaneshwar Shinde100% (2)
- Reviewer in Physical ScienceDocument3 pagesReviewer in Physical ScienceJayrick James AriscoNo ratings yet
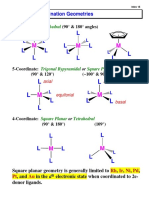
- Coordination Chemistry Theories and GeometriesDocument11 pagesCoordination Chemistry Theories and GeometriesZakiyah Rizkah Nu'manNo ratings yet
- Coordination CompoundsDocument43 pagesCoordination CompoundsVehlaNo ratings yet
- L-24 Cordination and CompundsDocument18 pagesL-24 Cordination and CompundsAkhilesh KumarNo ratings yet
- 2.2 - Introduction To Coordination Chemistry - IsomerismDocument47 pages2.2 - Introduction To Coordination Chemistry - IsomerismMphoNo ratings yet
- Coordinate Bonding Explained in 40 CharactersDocument8 pagesCoordinate Bonding Explained in 40 CharactersAnshuman TagoreNo ratings yet
- Coordinate Covalent Bond - WikipediaDocument3 pagesCoordinate Covalent Bond - Wikipediatsvmpm1765No ratings yet
- 1st Lecture On Co-Ordination CompoundsDocument14 pages1st Lecture On Co-Ordination CompoundsSharma Ji Ka BetaNo ratings yet
- Co Ordination 1Document33 pagesCo Ordination 1Nivi RajNo ratings yet
- Chemistry Exam Question2Document6 pagesChemistry Exam Question2akikoNo ratings yet
- Theory of Coordination CompoundsDocument30 pagesTheory of Coordination CompoundsDr. Md. Ehtesham Ul HoqueNo ratings yet
- XIICOORDINATIONModule 1Document7 pagesXIICOORDINATIONModule 1Arpit KumarNo ratings yet
- Coordination Chemistry: Werner's Theory, Chelate Effect, and Crystal Field TheoryDocument34 pagesCoordination Chemistry: Werner's Theory, Chelate Effect, and Crystal Field TheoryKuroko TetsuyaNo ratings yet
- chl9 (Sem 1 Inorganic)Document24 pageschl9 (Sem 1 Inorganic)rodrigues flemeNo ratings yet
- TEX - CHEM 103 Organic ChemistryDocument52 pagesTEX - CHEM 103 Organic ChemistrychioNo ratings yet
- WernerDocument5 pagesWernerSachin NagargojeNo ratings yet
- Synthesis and Characterization of Coordination CompoundsDocument13 pagesSynthesis and Characterization of Coordination CompoundsJ Mora GañanNo ratings yet
- History of Coordination CompoundsDocument18 pagesHistory of Coordination CompoundsIlmu Kimia CNo ratings yet
- Chemistry 2810 A Assignment AnswersDocument11 pagesChemistry 2810 A Assignment Answershodaps.ggsNo ratings yet
- Forces Between Atoms and MoleculesDocument13 pagesForces Between Atoms and MoleculesDoc_CrocNo ratings yet
- Chapter 15 - HydrocarbonsDocument16 pagesChapter 15 - HydrocarbonsNabindra RuwaliNo ratings yet
- BaiPhucChat-hinh (Compatibility Mode)Document49 pagesBaiPhucChat-hinh (Compatibility Mode)Thế anh PhạmNo ratings yet
- Solution Manual For Chemistry 9th EditionDocument36 pagesSolution Manual For Chemistry 9th Editionsaturantbruniontvg0100% (42)
- Full Download Solution Manual For Chemistry 9th Edition PDF Full ChapterDocument36 pagesFull Download Solution Manual For Chemistry 9th Edition PDF Full Chapterloudly.nereisnai6100% (19)
- Che QP 2018Document14 pagesChe QP 2018Vinay AdariNo ratings yet
- Ligand Subsitution On Carbonatopentamine Cobalt (III) NitrateDocument40 pagesLigand Subsitution On Carbonatopentamine Cobalt (III) Nitratevijaybenadict100% (2)
- Coordination Compounds PDFDocument107 pagesCoordination Compounds PDFMaria IslamNo ratings yet
- CBSE Class 12 Chemistry Paper Sample Paper Solution Set 4Document14 pagesCBSE Class 12 Chemistry Paper Sample Paper Solution Set 4Gamer ChannelNo ratings yet
- Synthesis and Color of Cr3+ ComplexesDocument18 pagesSynthesis and Color of Cr3+ ComplexesFelipe VilchesNo ratings yet
- 1 UNIT 5 (Nurul Inayah)Document18 pages1 UNIT 5 (Nurul Inayah)azizah.irwansyah27No ratings yet
- Solution Manual For Chemistry For Changing Times 14th Edition by Hill McCreary ISBN 0321972023 9780321972026Document36 pagesSolution Manual For Chemistry For Changing Times 14th Edition by Hill McCreary ISBN 0321972023 9780321972026lindseyallenizqtgdakmw100% (27)
- Carbocation - Wikipedia, The Free EncyclopediaDocument5 pagesCarbocation - Wikipedia, The Free EncyclopediaBenni WewokNo ratings yet
- Case in Point: Tetrahedral Vs Octahedral ComplexesDocument33 pagesCase in Point: Tetrahedral Vs Octahedral ComplexesShubham KumarNo ratings yet
- Creative Learning Classes, Karkala: Second Pu Annual Examination April - 2022 Chemistry Detailed SolutionDocument14 pagesCreative Learning Classes, Karkala: Second Pu Annual Examination April - 2022 Chemistry Detailed SolutionBazil 9393No ratings yet
- Angew Chem Int Ed Engl - October 1982 - Hoffmann - Building Bridges Between Inorganic and Organic Chemistry NobelDocument14 pagesAngew Chem Int Ed Engl - October 1982 - Hoffmann - Building Bridges Between Inorganic and Organic Chemistry NobelcalistaNo ratings yet
- Coordination Chemistry: A Guide to Structures, Isomers, and ReactivityDocument25 pagesCoordination Chemistry: A Guide to Structures, Isomers, and Reactivitypatel_346879839No ratings yet
- Coordination ChemistryDocument25 pagesCoordination Chemistryggwp21No ratings yet
- Answer For Physics 1 - 240318 - 192757Document26 pagesAnswer For Physics 1 - 240318 - 192757beharukassa10No ratings yet
- Langmuir 1916Document75 pagesLangmuir 1916Rose BleueNo ratings yet
- Coordination Chemistry: Werner's Theory and IUPAC NomenclatureDocument17 pagesCoordination Chemistry: Werner's Theory and IUPAC NomenclatureDipesh PanditNo ratings yet
- Slater-Pauling Behavior and Origin of The Half-Metallicity of The Full-Heusler Alloys PhysRevB.66.174429Document9 pagesSlater-Pauling Behavior and Origin of The Half-Metallicity of The Full-Heusler Alloys PhysRevB.66.174429rajanadarajanNo ratings yet
- Chem Project Term 1Document21 pagesChem Project Term 1Abishek ArunNo ratings yet
- Unit 2Document42 pagesUnit 2Muktaar HassenNo ratings yet
- Isomerism in Coordination CompoundsDocument15 pagesIsomerism in Coordination CompoundsRicky SharmaNo ratings yet
- Theories of Bonding in Coordination CompoundsDocument7 pagesTheories of Bonding in Coordination CompoundsDhanush KumarNo ratings yet
- Bonding and Hybridizatio1Document16 pagesBonding and Hybridizatio1AB AniketNo ratings yet
- Class 11 Chemistry Revision Notes Organic ChemistryDocument26 pagesClass 11 Chemistry Revision Notes Organic ChemistryNair SidharthNo ratings yet
- Javed Sceince Academy Composing 21-02-2017Document8 pagesJaved Sceince Academy Composing 21-02-2017Syed HasaniNo ratings yet
- Coordination Chemistry: Hui LiDocument91 pagesCoordination Chemistry: Hui LiPdssnNo ratings yet
- Transition Metal Handout (2018 - 04 - 16 01 - 41 - 52 UTC)Document9 pagesTransition Metal Handout (2018 - 04 - 16 01 - 41 - 52 UTC)patrice green - SteadmanNo ratings yet
- Solution Manual For General Chemistry 10th Edition Darrell D Ebbing Steven D GammonDocument36 pagesSolution Manual For General Chemistry 10th Edition Darrell D Ebbing Steven D Gammonvisearborist.af0eg100% (45)
- Google Analytics IQ Exam - Campaign Tracking QuestionsDocument4 pagesGoogle Analytics IQ Exam - Campaign Tracking QuestionsSwati GautamNo ratings yet
- Nehru and Civil Liberties PDFDocument6 pagesNehru and Civil Liberties PDFSwati GautamNo ratings yet
- Absorbance Vs Volume of Ni (II)Document1 pageAbsorbance Vs Volume of Ni (II)Swati GautamNo ratings yet
- For A Particular System It Was Found That 5 Const. 2: Problem 1Document3 pagesFor A Particular System It Was Found That 5 Const. 2: Problem 1Swati GautamNo ratings yet
- Alco ChemDocument15 pagesAlco Chemdeepthimahanthi sabithaNo ratings yet
- 10ml PDFDocument1 page10ml PDFSwati GautamNo ratings yet
- Fe(NO3)3 reaction rate over timeDocument1 pageFe(NO3)3 reaction rate over timeSwati GautamNo ratings yet
- SeqdumpDocument1 pageSeqdumpSwati GautamNo ratings yet
- BS Chemistry IitbombayDocument35 pagesBS Chemistry IitbombaySwati GautamNo ratings yet
- 2yr MSCDocument31 pages2yr MSCSwati GautamNo ratings yet
- Nptel: Protein Chemistry - Web CourseDocument3 pagesNptel: Protein Chemistry - Web CourseSwati GautamNo ratings yet
- Chemistry Books ListDocument136 pagesChemistry Books ListAlexander Martin13% (8)
- Co ComplexDocument13 pagesCo ComplexSwati GautamNo ratings yet
- Color of Transition Metal Complexes Explained by Electronic TransitionsDocument6 pagesColor of Transition Metal Complexes Explained by Electronic TransitionsSunreeta BhattacharyaNo ratings yet
- 111l Experiment 8 - NaohDocument10 pages111l Experiment 8 - NaohKrishna RajNo ratings yet
- ESC101: Introduction To Computing: Course LogisticsDocument23 pagesESC101: Introduction To Computing: Course LogisticsSwati GautamNo ratings yet
- Computational Drug Design Book ReviewDocument2 pagesComputational Drug Design Book ReviewSwati GautamNo ratings yet
- How to Write a Successful Application for NIH Research Training ProgramsDocument12 pagesHow to Write a Successful Application for NIH Research Training ProgramsCatherine ChengNo ratings yet
- Endsem BDocument13 pagesEndsem BSwati GautamNo ratings yet
- Spectro ChemicalDocument4 pagesSpectro ChemicalSwati GautamNo ratings yet
- Subject ChemistryDocument11 pagesSubject ChemistrySwati GautamNo ratings yet
- Thermodynamic Properties of an Ideal Monatomic GasDocument20 pagesThermodynamic Properties of an Ideal Monatomic GasradenbagusNo ratings yet
- Retrosynthetic Analysis PDFDocument6 pagesRetrosynthetic Analysis PDFHelmi FauziNo ratings yet
- 1 CHM616 Ligands Charges ElectronsDocument9 pages1 CHM616 Ligands Charges ElectronsSwati GautamNo ratings yet
- Spectro ChemicalDocument4 pagesSpectro ChemicalSwati GautamNo ratings yet
- Quino LinesDocument4 pagesQuino LinesSwati GautamNo ratings yet
- Analysis of Vitamin C: Linus Pauling Institute: The TitrationDocument5 pagesAnalysis of Vitamin C: Linus Pauling Institute: The TitrationSwati GautamNo ratings yet
- Name of The CourseDocument4 pagesName of The CourseSwati GautamNo ratings yet
- Quino LinesDocument4 pagesQuino LinesSwati GautamNo ratings yet
- BSMLS I - MT GravimetryDocument71 pagesBSMLS I - MT GravimetrynenaNo ratings yet
- Preparation of Rayon Thread by NadeemDocument3 pagesPreparation of Rayon Thread by NadeemRiyad KhanNo ratings yet
- Administrative Medical Assisting 8th Edition French Test BankDocument36 pagesAdministrative Medical Assisting 8th Edition French Test Bankpolymniahookerahd2c100% (51)
- Chemistry ArihantDocument31 pagesChemistry Arihantrahul100% (2)
- Chem41 - Isolation of LipidsDocument2 pagesChem41 - Isolation of LipidsSebastian SmytheNo ratings yet
- Patent Number Document Id Date Published: Process For Preparing Organic Fluorine CompoundsDocument6 pagesPatent Number Document Id Date Published: Process For Preparing Organic Fluorine CompoundsJyot VakhariaNo ratings yet
- Salt Analysis General ProcedureDocument7 pagesSalt Analysis General Procedurefranklin mahizhaNo ratings yet
- Ekstraksi Palladium Dari PCB Dengan Asam Nitrat - Solvent Ekstraksi Dan Precipitasi AmoniaDocument10 pagesEkstraksi Palladium Dari PCB Dengan Asam Nitrat - Solvent Ekstraksi Dan Precipitasi AmoniaAde SatriaNo ratings yet
- Heat of Chemical ReactionsDocument19 pagesHeat of Chemical ReactionsCikgu AnitaNo ratings yet
- 2224inst-Available Oia-1677 FIA (1) - CopiarDocument38 pages2224inst-Available Oia-1677 FIA (1) - CopiarFranco A. ZavaletaNo ratings yet
- GCSE Required Practical Tasks GuideDocument7 pagesGCSE Required Practical Tasks Guideastha patelNo ratings yet
- 2 Heat of PrecipitationDocument22 pages2 Heat of PrecipitationSyawal AnizamNo ratings yet
- Chapter Four: Reactions in Aqueous Solution: SolutionsDocument24 pagesChapter Four: Reactions in Aqueous Solution: SolutionsPaulAngeloPascuaNo ratings yet
- North Vista 2015 Prelim Paper 1Document20 pagesNorth Vista 2015 Prelim Paper 1GM MonsterEtaNo ratings yet
- GEA Lithium Battery Material Drying Spray Tcm11 34880Document10 pagesGEA Lithium Battery Material Drying Spray Tcm11 34880xianghuaokNo ratings yet
- CSEC Chemistry June 2005 P2Document16 pagesCSEC Chemistry June 2005 P2rampee charles100% (1)
- EXAMINER TIPS For Mastering IGCSE ChemistryDocument6 pagesEXAMINER TIPS For Mastering IGCSE ChemistryAnand Kumar100% (1)
- Chapter 16 - SaltsDocument3 pagesChapter 16 - SaltsschlemielzNo ratings yet
- EXPERIMENT 1 - Bendo Marjorie P.Document5 pagesEXPERIMENT 1 - Bendo Marjorie P.Bendo Marjorie P.100% (1)
- (I) Identification of Unorganized Crude Drugs - LabmonkDocument5 pages(I) Identification of Unorganized Crude Drugs - LabmonkKHUSHBOO KATHAROTIYANo ratings yet
- Important Question ICSE 2010 Class 10th Sulphur Dioxide Sulphuric Acid and Hydrogen SulphideDocument8 pagesImportant Question ICSE 2010 Class 10th Sulphur Dioxide Sulphuric Acid and Hydrogen SulphideYash KapoorNo ratings yet
- Test Bank For Sociology in Our Times 11th Edition Diana KendallDocument36 pagesTest Bank For Sociology in Our Times 11th Edition Diana Kendallzebrula.violatorzw1hf100% (34)
- Method Dna ExtractionDocument3 pagesMethod Dna ExtractionRayye Da ProNo ratings yet
- 2021 January (1C) QPDocument36 pages2021 January (1C) QPKevin MattNo ratings yet
- Structure 1.1 Preparation Agi Aqueous SolutionsDocument7 pagesStructure 1.1 Preparation Agi Aqueous SolutionsNubar MammadovaNo ratings yet
- Cambridge IGCSE: CHEMISTRY 0620/62Document12 pagesCambridge IGCSE: CHEMISTRY 0620/62ShehabNo ratings yet
- CH 35 TitrimetryDocument22 pagesCH 35 TitrimetrystupidbrowneyesNo ratings yet
- Occult Blood & Reducing SubstancesDocument54 pagesOccult Blood & Reducing SubstancesSULEIMAN OMARNo ratings yet
- t4 SC 1125 Aqa Gcse Chemistry Separate Science Unit 8 Chemical Analysis Knowledge Organiser - Ver - 5Document4 pagest4 SC 1125 Aqa Gcse Chemistry Separate Science Unit 8 Chemical Analysis Knowledge Organiser - Ver - 5S WintersNo ratings yet
- 2013 MirandaDocument93 pages2013 MirandaRazif YusufNo ratings yet