You might also like
- Fuel_Cells_Fundamentals_and_ApplicationsDocument35 pagesFuel_Cells_Fundamentals_and_ApplicationsManfredwangNo ratings yet
- Representing Vapor-Liquid Equilibrium For An Aqueous MEA-CO System Using The Electrolyte Nonrandom-Two-Liquid ModelDocument11 pagesRepresenting Vapor-Liquid Equilibrium For An Aqueous MEA-CO System Using The Electrolyte Nonrandom-Two-Liquid Modeldjc49No ratings yet
- Proton Exchange Fuel: CellsDocument12 pagesProton Exchange Fuel: CellsvlabdulazeemNo ratings yet
- Pavel Et Al-2014-Angewandte Chemie International Edition PDFDocument4 pagesPavel Et Al-2014-Angewandte Chemie International Edition PDFXav AguilasNo ratings yet
- Intro 8Document6 pagesIntro 8AlisaNo ratings yet
- Compound-Specific Imaging of Methanol Fuel Cell For Performance and Degradation Studies Using A Mini Positron Emission TomographDocument14 pagesCompound-Specific Imaging of Methanol Fuel Cell For Performance and Degradation Studies Using A Mini Positron Emission TomographAhmad RafiqanTB0009No ratings yet
- Nouveau Document Microsoft Office WordDocument11 pagesNouveau Document Microsoft Office WordZouaghi ZakariaNo ratings yet
- Climate Protection: in A NanospongeDocument8 pagesClimate Protection: in A NanospongeVipulNo ratings yet
- Design of Experiment To Predict The Time Between Hydrogen Purges For An Air-Breathing PEM Fuel Cell in Dead-End Mode in A Closed EnvironmentDocument12 pagesDesign of Experiment To Predict The Time Between Hydrogen Purges For An Air-Breathing PEM Fuel Cell in Dead-End Mode in A Closed EnvironmentChicheNo ratings yet
- Fuel Cell TypeDocument5 pagesFuel Cell TypeDarshann ParmarNo ratings yet
- Materials Today Physics: Tingting Wu, Wenjun Fan, Yang Zhang, Fuxiang ZhangDocument23 pagesMaterials Today Physics: Tingting Wu, Wenjun Fan, Yang Zhang, Fuxiang ZhangSelya AmandaNo ratings yet
- Isooctane in SOFCDocument5 pagesIsooctane in SOFCaymanoh056No ratings yet
- Revised ManuscriptDocument7 pagesRevised Manuscriptapi-3728640No ratings yet
- Benzimidazole As Solid Electrolyte Material For Fuel Cells: Daniel Herranz and Pilar OcónDocument21 pagesBenzimidazole As Solid Electrolyte Material For Fuel Cells: Daniel Herranz and Pilar OcónMiriam GarciaNo ratings yet
- Electrochemical Synthesis of Ammonia As ADocument4 pagesElectrochemical Synthesis of Ammonia As ADung Phan Thị ThùyNo ratings yet
- Microfluidic Fuel Cell Based On Laminar Flow: Eric R. Choban, Larry J. Markoski, Andrzej Wieckowski, Paul J.A. KenisDocument7 pagesMicrofluidic Fuel Cell Based On Laminar Flow: Eric R. Choban, Larry J. Markoski, Andrzej Wieckowski, Paul J.A. KenisEdwin Jhonatan Gachuz VazquezNo ratings yet
- A Powder Metallurgy Route To Produce Ni RaneyDocument8 pagesA Powder Metallurgy Route To Produce Ni RaneyLucas Vander MeyNo ratings yet
- Fuel Cell System and Their Technologies A ReviewDocument6 pagesFuel Cell System and Their Technologies A ReviewEditor IJTSRDNo ratings yet
- H 2 Production Via Ammonia Decomposition Using Non-Noble Metal Catalysts: A Review Bell - Et - Al-2016-Topics - in - Catalysis-VoRDocument20 pagesH 2 Production Via Ammonia Decomposition Using Non-Noble Metal Catalysts: A Review Bell - Et - Al-2016-Topics - in - Catalysis-VoRAr DiNo ratings yet
- Modeling of The Ballard-Mark-V Proton Exchange Membrane Fuel Cell With Power Converters For Applications in Autonomous Underwater VehiclesDocument14 pagesModeling of The Ballard-Mark-V Proton Exchange Membrane Fuel Cell With Power Converters For Applications in Autonomous Underwater VehiclesPeter VallejoNo ratings yet
- A Presentation On Distributed Power Generation Using Fuel CellsDocument12 pagesA Presentation On Distributed Power Generation Using Fuel Cellsapi-19799369No ratings yet
- Lecture 21 - Alternative Energy Resources - Fuel CellDocument5 pagesLecture 21 - Alternative Energy Resources - Fuel CellIbrar ahmadNo ratings yet
- Design and Simulation of A Liquid Electrolyte Passive Direct Methanol Fuel Cell With Low Methanol CrossoverDocument11 pagesDesign and Simulation of A Liquid Electrolyte Passive Direct Methanol Fuel Cell With Low Methanol CrossoverAlejandro Duvan Lopez RojasNo ratings yet
- Berning, Lu, Djilali - 2002 - Three-Dimensional Computational Analysis of Transport Phenomena in A PEM Fuel CellDocument11 pagesBerning, Lu, Djilali - 2002 - Three-Dimensional Computational Analysis of Transport Phenomena in A PEM Fuel CellMeita PratiwiNo ratings yet
- Vera PaperDocument7 pagesVera Paperمحمد أشرفNo ratings yet
- 2006 HH Meng Fuel CellDocument4 pages2006 HH Meng Fuel CellPrasanth KumarNo ratings yet
- Effect of Cathode Catalyst Layer Thickness On Methanol Cross-Over in A DMFCDocument7 pagesEffect of Cathode Catalyst Layer Thickness On Methanol Cross-Over in A DMFCKaustubhNo ratings yet
- Thiết Kế Máy - Huy TrầnDocument6 pagesThiết Kế Máy - Huy TrầnHuy TranNo ratings yet
- CO Poisoning - 1Document17 pagesCO Poisoning - 1Faseeh KKNo ratings yet
- PresentationonFuelCellsforStudents17thJune2012 PDFDocument38 pagesPresentationonFuelCellsforStudents17thJune2012 PDFYousef SailiniNo ratings yet
- Water-Gas Shift Mechanism on Pt/Ceria(111) Studied by DFTDocument12 pagesWater-Gas Shift Mechanism on Pt/Ceria(111) Studied by DFTleonardoNo ratings yet
- ECS Trans - 2015-Al-Musa-125-36Document12 pagesECS Trans - 2015-Al-Musa-125-36aymanoh056No ratings yet
- Chapter 1 6-19-07Document34 pagesChapter 1 6-19-07Richard WoudenbergNo ratings yet
- JPS Berningetal 2002Document12 pagesJPS Berningetal 2002GIOVANNI ACCARINONo ratings yet
- Photocatalytic Water Splitting With A Quantum Efficiency of Almost UnityDocument16 pagesPhotocatalytic Water Splitting With A Quantum Efficiency of Almost UnityNúria Ibars AlmirallNo ratings yet
- Bernt 2020 J. Electrochem. Soc. 167 124502Document13 pagesBernt 2020 J. Electrochem. Soc. 167 124502Aravind ShankarNo ratings yet
- Characteristics of Heat and Mass Transport in A Passive Direct Methanol Fuel Cell Operated With Concentrated MethanolDocument7 pagesCharacteristics of Heat and Mass Transport in A Passive Direct Methanol Fuel Cell Operated With Concentrated MethanolKaustubhNo ratings yet
- Lecture 21: Alternative Energy Resources - The Fuel CellDocument6 pagesLecture 21: Alternative Energy Resources - The Fuel CellIjazzzAliNo ratings yet
- dmfc387 PDFDocument9 pagesdmfc387 PDFKaustubhNo ratings yet
- Alkaline Fuel Cell Design PrinciplesDocument9 pagesAlkaline Fuel Cell Design PrinciplesPooveanthan HbNo ratings yet
- Article J of Power SourcesDocument14 pagesArticle J of Power SourcesMejdoubNo ratings yet
- articolo AnnaDocument9 pagesarticolo AnnaDiego MaportiNo ratings yet
- Fuel Cells: Do You Know? What Is A Fuel Cell?Document4 pagesFuel Cells: Do You Know? What Is A Fuel Cell?manish kumarNo ratings yet
- Efchp Fuelcell23Document2 pagesEfchp Fuelcell23Md Raisul IslamNo ratings yet
- Jcice 64Document7 pagesJcice 64api-3728640No ratings yet
- A Micro Methanol Fuel Cell Operating at Near Room TemperatureDocument4 pagesA Micro Methanol Fuel Cell Operating at Near Room TemperaturePrasanth KumarNo ratings yet
- Noik 2006Document12 pagesNoik 2006Adryan MarstyaNo ratings yet
- 10.1016@S1359 02860200108 0Document11 pages10.1016@S1359 02860200108 0Meita PratiwiNo ratings yet
- Fuel Cell PPT EP452Document22 pagesFuel Cell PPT EP452Dhruv BawejaNo ratings yet
- ModelAFC PublDocument11 pagesModelAFC PublJacob WilsonNo ratings yet
- High Activity Cobalt Based Catalysts For The Carbonylation of MethanolDocument2 pagesHigh Activity Cobalt Based Catalysts For The Carbonylation of MethanolSUSINo ratings yet
- Efficiency and Consistency Enhancement For Alkaline Electrolyzers Driven by Renewable Energy SourcesDocument13 pagesEfficiency and Consistency Enhancement For Alkaline Electrolyzers Driven by Renewable Energy Sourcessde goonNo ratings yet
- Fuel CellDocument4 pagesFuel CellTilak Raj RaiNo ratings yet
- 1 s2.0 S0360319921005115 MainDocument14 pages1 s2.0 S0360319921005115 MainPatrice PariNo ratings yet
- Industrial Aluminium Production The Hall-Heroult PDocument11 pagesIndustrial Aluminium Production The Hall-Heroult Pmamun.du027No ratings yet
- Chlor AlkaliDocument15 pagesChlor AlkaliDaniela BagashovskaNo ratings yet
- Report On "Study Ofdirect Methanol Fuel Cell": Technical Report and Seminar (TRS)Document10 pagesReport On "Study Ofdirect Methanol Fuel Cell": Technical Report and Seminar (TRS)Somnath MaityNo ratings yet
- Review of Methane Catalytic Cracking For Hydrogen ProductionDocument32 pagesReview of Methane Catalytic Cracking For Hydrogen ProductionVishal GoswamiNo ratings yet
- Biohydrogen III: Renewable Energy System by Biological Solar Energy ConversionFrom EverandBiohydrogen III: Renewable Energy System by Biological Solar Energy ConversionNo ratings yet
- Biofilms in Bioelectrochemical Systems: From Laboratory Practice to Data InterpretationFrom EverandBiofilms in Bioelectrochemical Systems: From Laboratory Practice to Data InterpretationNo ratings yet
- Bio-jet Fuel Conversion Technologies ReviewDocument24 pagesBio-jet Fuel Conversion Technologies Reviewaisyahzafira26No ratings yet
- Ameen 2017Document17 pagesAmeen 2017aisyahzafira26No ratings yet
- Arun 2015Document16 pagesArun 2015aisyahzafira26No ratings yet
- Lin 2020Document9 pagesLin 2020aisyahzafira26No ratings yet
- Ameen 2019Document9 pagesAmeen 2019aisyahzafira26No ratings yet
- Molecular Catalysis: B. Pawelec, C.V. Loricera, C. Geantet, N. Mota, J.L.G. Fierro, R.M. NavarroDocument9 pagesMolecular Catalysis: B. Pawelec, C.V. Loricera, C. Geantet, N. Mota, J.L.G. Fierro, R.M. Navarroaisyahzafira26No ratings yet
- Sotelo 2011Document9 pagesSotelo 2011aisyahzafira26No ratings yet
- Molecular Catalysis: B. Pawelec, C.V. Loricera, C. Geantet, N. Mota, J.L.G. Fierro, R.M. NavarroDocument9 pagesMolecular Catalysis: B. Pawelec, C.V. Loricera, C. Geantet, N. Mota, J.L.G. Fierro, R.M. Navarroaisyahzafira26No ratings yet
- Solvent ImportanceDocument6 pagesSolvent ImportanceAbhiNo ratings yet
- What Is A Direct Methanol Fuel CellDocument25 pagesWhat Is A Direct Methanol Fuel Cellaisyahzafira26No ratings yet
- Sythesis of Bioavture Through Hydrodeoxygenation and Catalytic Cracking From Oleic Acid Using Nimo/Zeolit CatalystDocument5 pagesSythesis of Bioavture Through Hydrodeoxygenation and Catalytic Cracking From Oleic Acid Using Nimo/Zeolit Catalystaisyahzafira26No ratings yet
- 2005 Rev Pre FlashDocument5 pages2005 Rev Pre Flashghasem_726990287No ratings yet
- Hamnet T 1997Document13 pagesHamnet T 1997aisyahzafira26No ratings yet
- Understanding CatalysisDocument34 pagesUnderstanding Catalysisaisyahzafira26No ratings yet
- Zea2005 PDFDocument9 pagesZea2005 PDFaisyahzafira26No ratings yet
- Skripsi UI Fischer TropschDocument17 pagesSkripsi UI Fischer Tropschaisyahzafira26No ratings yet
- Hydrocracking of Isoheptadecanes On Pt/H-ZSM-22: An Example of Pore Mouth CatalysisDocument8 pagesHydrocracking of Isoheptadecanes On Pt/H-ZSM-22: An Example of Pore Mouth Catalysisaisyahzafira26No ratings yet
- IPB Fischer TropschDocument10 pagesIPB Fischer Tropschaisyahzafira26No ratings yet
- Aa00000383 00143 00210Document6 pagesAa00000383 00143 00210Adinda Asri PixelinaNo ratings yet
- Farmasi FisikDocument4 pagesFarmasi Fisikyosafat mustikoartoNo ratings yet
- Mineline MSP Sell SheetDocument2 pagesMineline MSP Sell SheetMary Huaylla ANo ratings yet
- Reaction PaperDocument3 pagesReaction PaperCamilla Reyes100% (1)
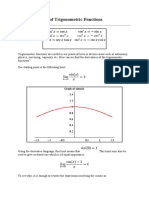
- The Derivatives of Trigonometric FunctionsDocument29 pagesThe Derivatives of Trigonometric FunctionsM Arifin RasdhakimNo ratings yet
- Wells Fargo Preferred CheckingDocument4 pagesWells Fargo Preferred Checkingjames50% (2)
- It Complaint Management SystemDocument26 pagesIt Complaint Management SystemKapil GargNo ratings yet
- TCS Case StudyDocument21 pagesTCS Case StudyJahnvi Manek0% (1)
- Germination DiscussionDocument3 pagesGermination DiscussionMay Lacdao100% (1)
- 3126B Industrial Engine Electrical System: ECM AMP ConnectorsDocument2 pages3126B Industrial Engine Electrical System: ECM AMP Connectorsedcar100% (1)
- TDS - J TOWhead D60Document1 pageTDS - J TOWhead D60TahirNo ratings yet
- 3D MapsDocument1,128 pages3D MapsjoangopanNo ratings yet
- Characterization of uPVC Pipes Using Thermal AnalysisDocument7 pagesCharacterization of uPVC Pipes Using Thermal AnalysisFaiz SabianNo ratings yet
- Thursday: Dhaka Electric Supply Company Limited (DESCO) Load Shedding Schedule On 11 KV FeedersDocument16 pagesThursday: Dhaka Electric Supply Company Limited (DESCO) Load Shedding Schedule On 11 KV FeedersaajahidNo ratings yet
- Minimal Preludes I & II - Jeroen Van VeenDocument5 pagesMinimal Preludes I & II - Jeroen Van VeenErnesto HartmannNo ratings yet
- Propoxur (WHO Pesticide Residues Series 3)Document26 pagesPropoxur (WHO Pesticide Residues Series 3)Desye MeleseNo ratings yet
- Week 10 - PROG 8510 Week 10Document16 pagesWeek 10 - PROG 8510 Week 10Vineel KumarNo ratings yet
- Glenn GreenbergDocument3 pagesGlenn Greenbergannsusan21No ratings yet
- RAM TP Report 2021Document15 pagesRAM TP Report 2021Sowmya AnanthaNo ratings yet
- 2.1.3.a EthicsSafetyDocument3 pages2.1.3.a EthicsSafetyBenNo ratings yet
- ARNU15GL2G4Document2 pagesARNU15GL2G4julioNo ratings yet
- Hospitality Enterprise 2 RajendraDocument31 pagesHospitality Enterprise 2 RajendraChristian AgultoNo ratings yet
- A Paper On Cactaceae, Lobivia Ferox A New Record in Chilean FloraDocument8 pagesA Paper On Cactaceae, Lobivia Ferox A New Record in Chilean FloraJohn GamesbyNo ratings yet
- MODULE 7-11 Notes PrefiDocument7 pagesMODULE 7-11 Notes PrefiPASCUAL, ALJON R.No ratings yet
- CCGNHS Ste Stem SB Resolution 2023Document7 pagesCCGNHS Ste Stem SB Resolution 2023Rhodail Andrew CastroNo ratings yet
- Biology of KundaliniDocument591 pagesBiology of KundaliniMaureen Shoe100% (24)
- CASR PART 43 Amdt. 1 PDFDocument21 pagesCASR PART 43 Amdt. 1 PDFarbypratamax66100% (1)
- s7 1500 Compare Table en MnemoDocument88 pagess7 1500 Compare Table en MnemoPeli JorroNo ratings yet
- Debpriyo Roy Lab 1Document7 pagesDebpriyo Roy Lab 1udaikiranNo ratings yet
- Chapter 1Document19 pagesChapter 1Shehzana MujawarNo ratings yet
- Software Verification: AISC 360-16 SFD Example 001Document7 pagesSoftware Verification: AISC 360-16 SFD Example 001Dompu MengajiNo ratings yet
- Well Logging Interpretation Methodology For Carbonate Formation Fracture System Properties DeterminationDocument11 pagesWell Logging Interpretation Methodology For Carbonate Formation Fracture System Properties Determinationel hadiNo ratings yet