You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Why I Left Orthodox MedicineDocument179 pagesWhy I Left Orthodox MedicinejhNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Dopaminergic Pathways - LAGDocument30 pagesDopaminergic Pathways - LAGalvinbb100% (1)
- Clinical Investigations - On The Move - 2012 - by Tamer SolimanDocument268 pagesClinical Investigations - On The Move - 2012 - by Tamer Solimanyousrazeidan1979No ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Cancer Treatment TypesDocument43 pagesCancer Treatment TypesM Sai Pradhyumna100% (2)
- MED SURG FINAL EXAM QUESTION BANKDocument16 pagesMED SURG FINAL EXAM QUESTION BANKamelis100% (4)
- Approach To The Adult With Unexplained Pancytopenia - UpToDateDocument28 pagesApproach To The Adult With Unexplained Pancytopenia - UpToDatesusanaNo ratings yet
- Toxic Responses of The BloodDocument10 pagesToxic Responses of The BloodM. Joyce100% (2)
- Ascp Pointers MicroDocument73 pagesAscp Pointers Microraica56_362842087100% (5)
- Dass 21Document2 pagesDass 21J Iván FernándezNo ratings yet
- Genome Poster 2009Document1 pageGenome Poster 2009Öznur ErdoğanNo ratings yet
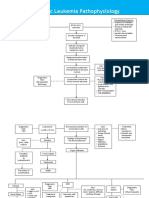
- Acute Lymphoblastic Leukemia Pathophysiology: Predisposing Factors: Etiology: Precipitating FactorsDocument3 pagesAcute Lymphoblastic Leukemia Pathophysiology: Predisposing Factors: Etiology: Precipitating FactorsKyla ValenciaNo ratings yet
- Maternal LecDocument23 pagesMaternal LecKate Onniel Rimando100% (1)
- NCP LeukemiaDocument3 pagesNCP LeukemiaLuige Avila100% (5)
- Child Health NSG Important QuestionsDocument10 pagesChild Health NSG Important Questionsedrinsne100% (2)
- Clinical Case in Neurology by SlidesgoDocument46 pagesClinical Case in Neurology by SlidesgoeaeapewNo ratings yet
- Rosenberg Self-Esteem ScaleDocument2 pagesRosenberg Self-Esteem ScaleGeorge WinchesterNo ratings yet
- Abnormal Uterine BleedingDocument19 pagesAbnormal Uterine BleedingGeorge WinchesterNo ratings yet
- Abnormal Uterine BleedingDocument19 pagesAbnormal Uterine BleedingGeorge WinchesterNo ratings yet
- Arterial Blood Gases JournalDocument5 pagesArterial Blood Gases JournalEllehcor BenwisanNo ratings yet
- SepsisDocument30 pagesSepsisGeorge WinchesterNo ratings yet
- Ability of Procalcitonin To Predict Bacterial MeningitisDocument20 pagesAbility of Procalcitonin To Predict Bacterial MeningitisGeorge WinchesterNo ratings yet
- BPPV Compared To Idiopathic BPPV and BPPV Secondary To Vestibular Neuritis (VN) PDFDocument4 pagesBPPV Compared To Idiopathic BPPV and BPPV Secondary To Vestibular Neuritis (VN) PDFGeorge WinchesterNo ratings yet
- Laryngopharyngealreflux2 120729003547 Phpapp01 PDFDocument25 pagesLaryngopharyngealreflux2 120729003547 Phpapp01 PDFGeorge WinchesterNo ratings yet
- Neuro-Ophthalmic Disorder Cranial NerveDocument4 pagesNeuro-Ophthalmic Disorder Cranial NerveGeorge WinchesterNo ratings yet
- Tropmed 22 Feb 2018 StudentDocument69 pagesTropmed 22 Feb 2018 StudentGeorge WinchesterNo ratings yet
- READDocument1 pageREADGeorge WinchesterNo ratings yet
- Lapkas RM Stroke FixDocument12 pagesLapkas RM Stroke FixGeorge WinchesterNo ratings yet
- Lymphatic Filariasis: Tropical Medicine BlockDocument26 pagesLymphatic Filariasis: Tropical Medicine BlockGeorge WinchesterNo ratings yet
- Pathogenesis of DKA and HHSDocument3 pagesPathogenesis of DKA and HHSGeorge WinchesterNo ratings yet
- Multidimensional Scale of Perceived Social Support: RequiredDocument2 pagesMultidimensional Scale of Perceived Social Support: RequiredGeorge WinchesterNo ratings yet
- Relations Between Perfectionism, Self-Esteem and Depression in Late AdolescentsDocument14 pagesRelations Between Perfectionism, Self-Esteem and Depression in Late AdolescentsHeru Imron KhoiriNo ratings yet
- Mental Health Status of Adolescents in South-East Asia:: Evidence For ActionDocument84 pagesMental Health Status of Adolescents in South-East Asia:: Evidence For ActionGeorge WinchesterNo ratings yet
- Table LeukimiaDocument3 pagesTable LeukimiaGeorge WinchesterNo ratings yet
- Diagnostico y Tratamiento de La Diabetes Tipo II ADA 2014Document67 pagesDiagnostico y Tratamiento de La Diabetes Tipo II ADA 2014caluca1987No ratings yet
- Content ServerDocument18 pagesContent ServerGeorge WinchesterNo ratings yet
- Diabetes Mellitus Ada 2014 Standards of Medical Care in Diabetes Diabetes Care 1.2014Document67 pagesDiabetes Mellitus Ada 2014 Standards of Medical Care in Diabetes Diabetes Care 1.2014George WinchesterNo ratings yet
- Quiz 2 CTB Epid Sept 08 2014 With AnswerDocument3 pagesQuiz 2 CTB Epid Sept 08 2014 With AnswerGeorge WinchesterNo ratings yet
- Coba Hitung Sampel SkripsiDocument12 pagesCoba Hitung Sampel SkripsiGeorge WinchesterNo ratings yet
- Self Measures For Self-Esteem Rosenberg Self-EsteemDocument4 pagesSelf Measures For Self-Esteem Rosenberg Self-Esteemapi-349021587No ratings yet
- Self Esteem ScaleDocument1 pageSelf Esteem ScaleGeorge WinchesterNo ratings yet
- The Influence of Gender On Social Anxiety Spectrum Symptoms in A Sample of University StudentsDocument7 pagesThe Influence of Gender On Social Anxiety Spectrum Symptoms in A Sample of University StudentsGeorge WinchesterNo ratings yet
- Acute Lymphocytic LeukemiaDocument7 pagesAcute Lymphocytic LeukemiaKim Enrico JumarangNo ratings yet
- Sobrevida Niños Cancer ImssDocument9 pagesSobrevida Niños Cancer ImssChristian Escalante DelgadoNo ratings yet
- VivaDocument3 pagesVivaMythily VedhagiriNo ratings yet
- Myeloproliferative DisorderDocument36 pagesMyeloproliferative DisorderKalpana ShahNo ratings yet
- Incidence of Hematologic Malignancies in EuropeDocument11 pagesIncidence of Hematologic Malignancies in EuropegiulioNo ratings yet
- Acute Myeloid Leukemia PathophysiologyDocument6 pagesAcute Myeloid Leukemia PathophysiologyChristine CollesNo ratings yet
- Doxotil BrochurDocument2 pagesDoxotil BrochurAnonymous cxfknMNo ratings yet
- RBC Disorders, Bleeding Disorders, and Pharmacology ReviewDocument11 pagesRBC Disorders, Bleeding Disorders, and Pharmacology ReviewSyed Muhammad HameemNo ratings yet
- Tumour Inhibitors From PlantsDocument59 pagesTumour Inhibitors From PlantsAlishba MushtaqNo ratings yet
- LeukemiareviewarticleDocument18 pagesLeukemiareviewarticleMansfield PharmaNo ratings yet
- Functions of Blood and ComponentsDocument15 pagesFunctions of Blood and ComponentsayuNo ratings yet
- Hematological Disorders in Geriatric PatientsDocument18 pagesHematological Disorders in Geriatric PatientsAndre HawkNo ratings yet
- Afp 20140501 P 731Document8 pagesAfp 20140501 P 731metabolismeproteinNo ratings yet
- Journal of Infection and Public Health: Tanzila SabaDocument16 pagesJournal of Infection and Public Health: Tanzila SabaprnjanNo ratings yet
- Leukemias: Care SettingDocument11 pagesLeukemias: Care SettingTinNo ratings yet
- Nucleic AcidsDocument66 pagesNucleic Acidspjblk100% (1)
- Chronic Lymphocytic Leukemia Present StatusDocument18 pagesChronic Lymphocytic Leukemia Present StatusLhOi ParagasNo ratings yet
- Leukemia Lesson PlanDocument5 pagesLeukemia Lesson PlanTopeshwar TpkNo ratings yet