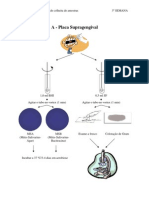
You might also like
- PROGORALDocument36 pagesPROGORALapi-26429188No ratings yet
- OralDocument54 pagesOralapi-26429188100% (2)
- Microbiologia Oral Esquema Protocolo 10 - SEMANA API20ADocument7 pagesMicrobiologia Oral Esquema Protocolo 10 - SEMANA API20Aapi-26429188No ratings yet
- FW Micro Oral Micro-2 - FreqDocument1 pageFW Micro Oral Micro-2 - Freqapi-26429188No ratings yet
- Examemicrobiologiajulho 2005Document1 pageExamemicrobiologiajulho 2005api-26429188No ratings yet
- Microbiologia Oral 3 AULA TPDocument13 pagesMicrobiologia Oral 3 AULA TPapi-26429188100% (1)
- 2 Frekmicrobiologia2005Document1 page2 Frekmicrobiologia2005api-26429188No ratings yet
- HepatiteDocument3 pagesHepatiteapi-26429188No ratings yet
- HepatiteDocument3 pagesHepatiteapi-26429188No ratings yet
- Doenças PeriodontaisDocument6 pagesDoenças Periodontaisapi-26429188100% (3)
- Microbiologia Oral Microbiologia Oral ApontamentosDocument6 pagesMicrobiologia Oral Microbiologia Oral Apontamentosapi-26429188100% (2)
- 2 Frekmicrobiologia2005Document1 page2 Frekmicrobiologia2005api-26429188No ratings yet
- Esquema - Protocolo 3 SemanaDocument3 pagesEsquema - Protocolo 3 Semanaapi-26429188No ratings yet
- Exame de Micro - Fev2004Document6 pagesExame de Micro - Fev2004api-26429188No ratings yet
- Ex Ames 1Document13 pagesEx Ames 1api-26429188No ratings yet
- BactériasDocument2 pagesBactériasapi-26429188No ratings yet
- Exame de Micro Geral - Fevereiro de 2005 MDDocument8 pagesExame de Micro Geral - Fevereiro de 2005 MDapi-26429188No ratings yet
- Esquema Protocolo 5 SEMANADocument7 pagesEsquema Protocolo 5 SEMANAapi-26429188No ratings yet
- Doenças PeriodontaisDocument6 pagesDoenças Periodontaisapi-26429188100% (3)
- Microbiologia Oral Esquema Protocolo 10 - SEMANA API20ADocument7 pagesMicrobiologia Oral Esquema Protocolo 10 - SEMANA API20Aapi-26429188No ratings yet
- Escolha Multipla MICRO ORAL 1º Teste PraticoDocument2 pagesEscolha Multipla MICRO ORAL 1º Teste Praticoapi-26429188No ratings yet
- Esquema Protocolo 4 SEMANADocument3 pagesEsquema Protocolo 4 SEMANAapi-26429188No ratings yet
- Escolha Multipla MICRO ORAL 1º Teste PraticoDocument2 pagesEscolha Multipla MICRO ORAL 1º Teste Praticoapi-26429188No ratings yet
- CarieDocument5 pagesCarieapi-26429188No ratings yet
- Escolha Multipla MICRO ORAL 1º Teste PraticoDocument2 pagesEscolha Multipla MICRO ORAL 1º Teste Praticoapi-26429188No ratings yet
- 3 Aula TPDocument23 pages3 Aula TPapi-26429188No ratings yet
- Doenças PeriodontaisDocument6 pagesDoenças Periodontaisapi-26429188100% (3)
- 2 Freq MicroDocument5 pages2 Freq Microapi-26429188No ratings yet
- Microbiologia Oral Microbiologia Oral ApontamentosDocument6 pagesMicrobiologia Oral Microbiologia Oral Apontamentosapi-26429188100% (2)
- 2 Freq MicroDocument5 pages2 Freq Microapi-26429188No ratings yet
- Orientações Técnicas para o PAIF - Vol. 1Document84 pagesOrientações Técnicas para o PAIF - Vol. 1Vinicius CescaNo ratings yet
- Resolução de problemas de funções e logaritmosDocument61 pagesResolução de problemas de funções e logaritmosEdielson HernandesNo ratings yet
- FichaDocument2 pagesFichaAlexanderPalerme67% (3)
- Direito Administrativo - Liçoes IntroduçaoDocument663 pagesDireito Administrativo - Liçoes IntroduçaoAnonymous O73A0EwmSCNo ratings yet
- Apresentaçao Ciencias Dos MateriaisDocument10 pagesApresentaçao Ciencias Dos MateriaisAndrielly CarolineNo ratings yet
- Rafael Luglio Guedes DEFINITIVO 3105Document68 pagesRafael Luglio Guedes DEFINITIVO 3105rafael guedesNo ratings yet
- Manual Dos Exames CardíacosDocument45 pagesManual Dos Exames CardíacosMatheus BaraldiNo ratings yet
- Lista de Exercícios 02 – Aritmética BináriaDocument2 pagesLista de Exercícios 02 – Aritmética BináriaTeo JavaNo ratings yet
- Segurança diária e meio ambiente em discussãoDocument14 pagesSegurança diária e meio ambiente em discussãoSinalize SetNo ratings yet
- Guia Do Programador Joel SaadeDocument9 pagesGuia Do Programador Joel SaadeWellington José CavalcantiNo ratings yet
- Slide Cálculos Químicos e EstequiométricosDocument44 pagesSlide Cálculos Químicos e Estequiométricoslidiacarolineferreira.silva2No ratings yet
- 7 ErrosDocument20 pages7 Errosrenatamruiz100% (3)
- Cravo Da Índia - Seus Poderes Mágicos - Oficina PDFDocument4 pagesCravo Da Índia - Seus Poderes Mágicos - Oficina PDFElton Castorino100% (2)
- Todos os benefícios da Moringa para a saúdeDocument4 pagesTodos os benefícios da Moringa para a saúdeJosé Canamala PhiriNo ratings yet
- Pisa 2012Document23 pagesPisa 2012Denizete Mesquita MesquitaNo ratings yet
- Práticas baseadas em evidências na PsicologiaDocument27 pagesPráticas baseadas em evidências na PsicologiaChristiano RodriguesNo ratings yet
- Equinócios e PáscoasDocument78 pagesEquinócios e PáscoasMário Renato MarianoNo ratings yet
- Ctic9 - MC - E3 - Geral DigestãoDocument1 pageCtic9 - MC - E3 - Geral DigestãoSilvinhoCostaNo ratings yet
- Prova - Cfs 1 2021 - Cod - 33Document28 pagesProva - Cfs 1 2021 - Cod - 33Desqueronee XesqueroneeNo ratings yet
- Legislacao e Normatizacao Aplicada Unid.1Document64 pagesLegislacao e Normatizacao Aplicada Unid.1Italo HugoNo ratings yet
- Vale Da Estranheza (Anna Wiener)Document246 pagesVale Da Estranheza (Anna Wiener)Antonio AndradeNo ratings yet
- Trabalho Prático Levantamento de Requisitos Sistema Entrega de Trabalhos EscolaresDocument43 pagesTrabalho Prático Levantamento de Requisitos Sistema Entrega de Trabalhos EscolaresRodrigo MartinsNo ratings yet
- Comportamento Organizacional e Liderança EficazDocument22 pagesComportamento Organizacional e Liderança EficazJadson CunhaNo ratings yet
- Livro Sistemas BiologicosDocument133 pagesLivro Sistemas BiologicosJESSICA FERNANDES REGUERA RUIZ100% (1)
- Exercícios resolvidos de juros simples e compostosDocument3 pagesExercícios resolvidos de juros simples e compostosAnonymous CZhW58yH0100% (2)
- Código de Falhas InverterDocument2 pagesCódigo de Falhas InverterAlchemist7No ratings yet
- Como conquistar a felicidade com a TRECDocument112 pagesComo conquistar a felicidade com a TRECRita De Cassia MathaisNo ratings yet
- Mielopatia DegenerativaDocument3 pagesMielopatia Degenerativasara ferreiraNo ratings yet
- Revista Cultura Do Automóvel - Ed. 35 - Maio22Document52 pagesRevista Cultura Do Automóvel - Ed. 35 - Maio22alemao7x1 DiehlNo ratings yet
- Brasil Belgica PDFDocument380 pagesBrasil Belgica PDFFernanda BeuxNo ratings yet