You might also like
- Gaussian 09 User's ReferenceDocument431 pagesGaussian 09 User's ReferenceWilliamNo ratings yet
- Griffiths D.J. Introduction To Quantum Mechanics (PH, 1995) (T) (408s)Document408 pagesGriffiths D.J. Introduction To Quantum Mechanics (PH, 1995) (T) (408s)BEATRIZ JAIMES GARCIA100% (1)
- Frontiers Quantum Chemsitry PDFDocument507 pagesFrontiers Quantum Chemsitry PDFRamón López FernándezNo ratings yet
- Electronic Structure TheoryDocument46 pagesElectronic Structure TheoryMAVERICK_HUNTER1234936No ratings yet
- Crystal Field Theory, Tight-Binding Method and Jahn-Teller EffectDocument39 pagesCrystal Field Theory, Tight-Binding Method and Jahn-Teller Effectdragonlj1No ratings yet
- Quantum Chemistry With Jaguar For Beginners: in This GuideDocument21 pagesQuantum Chemistry With Jaguar For Beginners: in This GuideChristian Mark Labiano100% (1)
- Basis SetsDocument28 pagesBasis SetsDipesh SomvanshiNo ratings yet
- Basissets PDFDocument19 pagesBasissets PDFYewula Debre Medhanit KidanemihiretNo ratings yet
- ElectronicStructure 3rdDocument23 pagesElectronicStructure 3rdAnonymous rn2qoBPjKyNo ratings yet
- Introduction To Electron CorrelationDocument37 pagesIntroduction To Electron CorrelationAnish RaoNo ratings yet
- Quantum Chapter 8 2016Document17 pagesQuantum Chapter 8 2016Aprajita PaliwalNo ratings yet
- Lec 8Document2 pagesLec 8jaurav41No ratings yet
- Basis Sets and PseudopotentialsDocument34 pagesBasis Sets and PseudopotentialssouvenirsouvenirNo ratings yet
- DFTDocument36 pagesDFTAnanth RaghavNo ratings yet
- Atomic Orbital Basis SetsDocument23 pagesAtomic Orbital Basis SetsJosé CortésNo ratings yet
- 06 Metode Ab InitioDocument26 pages06 Metode Ab InitioAbid Al Chem NugamaNo ratings yet
- Fundamentals of Molecular Integrals EvaluationDocument37 pagesFundamentals of Molecular Integrals EvaluationMarko MitićNo ratings yet
- MIT8 851S13 IntroToSCETDocument10 pagesMIT8 851S13 IntroToSCETalspeer1905No ratings yet
- 4 Molecular Orbital MethodsDocument40 pages4 Molecular Orbital MethodstudoorNo ratings yet
- Huckel Molecular Orbital Theory: Sapan Kumar Jain Assistant Professor, JMIDocument45 pagesHuckel Molecular Orbital Theory: Sapan Kumar Jain Assistant Professor, JMIMayank GuptNo ratings yet
- 7 - The MethodsDocument14 pages7 - The MethodsludihemicarNo ratings yet
- Homework 6Document2 pagesHomework 6ludihemicarNo ratings yet
- Edward Valeev - Basis Sets in Quantum ChemistryDocument26 pagesEdward Valeev - Basis Sets in Quantum ChemistryElectro_LiteNo ratings yet
- Basics of Quantum ChemistryDocument36 pagesBasics of Quantum ChemistryPace RaditNo ratings yet
- Fault-Tolerant Quantum Computation by AnyonsDocument27 pagesFault-Tolerant Quantum Computation by Anyonsfarsad ahmadNo ratings yet
- Barnett - The Calculation of Spherical Bessel Functions and Coulomb FunctionsDocument19 pagesBarnett - The Calculation of Spherical Bessel Functions and Coulomb FunctionszvirkoNo ratings yet
- Physics 195ab Course Notes 020917 F. PorterDocument705 pagesPhysics 195ab Course Notes 020917 F. Porterkevinchu021195No ratings yet
- Self-Consistent FieldDocument6 pagesSelf-Consistent FieldmekokiNo ratings yet
- Diodelaser: Prof. Dr.-Ing. DickmannDocument26 pagesDiodelaser: Prof. Dr.-Ing. DickmannАндрей АндреевNo ratings yet
- Fault-Tolerant Quantum Computation by Anyons: A.Yu. KitaevDocument29 pagesFault-Tolerant Quantum Computation by Anyons: A.Yu. KitaevNomentsuNo ratings yet
- Lec 1Document3 pagesLec 1laksh688No ratings yet
- Tiny Quater Ni On Dynamics Library 006Document71 pagesTiny Quater Ni On Dynamics Library 006Brian BeckmanNo ratings yet
- Surrey Mini-School R. F. CastenDocument41 pagesSurrey Mini-School R. F. CastenSdocNo ratings yet
- Kronig PenneyDocument25 pagesKronig PenneyAbel Tom100% (1)
- 10 Self-consistent field theory: ² ψ − ∇ ψ dr - ψ - r − r - ψ δ dr ψ - r − r - ψDocument4 pages10 Self-consistent field theory: ² ψ − ∇ ψ dr - ψ - r − r - ψ δ dr ψ - r − r - ψursml12No ratings yet
- Statistical Mechanics Lecture Notes (2006), L16Document4 pagesStatistical Mechanics Lecture Notes (2006), L16OmegaUserNo ratings yet
- Introduction To The Theory of Pseudopotentials: Patrick BriddonDocument43 pagesIntroduction To The Theory of Pseudopotentials: Patrick BriddonSiddheshwar ChopraNo ratings yet
- 1) Spin-Polarised Calculations 2) Geometry Optimisation: CHEM6085: Density Functional TheoryDocument14 pages1) Spin-Polarised Calculations 2) Geometry Optimisation: CHEM6085: Density Functional TheorylotannaNo ratings yet
- Introduction To String TheoryDocument85 pagesIntroduction To String TheoryFarhad HossainNo ratings yet
- Schrodinger: Hydrogen Helium Lithium Beryllium SodiumDocument2 pagesSchrodinger: Hydrogen Helium Lithium Beryllium Sodiumrujean romy p guisandoNo ratings yet
- EUE - From Diatomics To Graphene - 2016 PDFDocument56 pagesEUE - From Diatomics To Graphene - 2016 PDFluzanovNo ratings yet
- Cole HydrogenDocument15 pagesCole HydrogenDyra KesumaNo ratings yet
- Gaussian Basis Sets: CHEM6085: Density Functional TheoryDocument21 pagesGaussian Basis Sets: CHEM6085: Density Functional TheoryThiago MenzonattoNo ratings yet
- Inverse Kinematics Algorithms for Protein Structure DeterminationDocument13 pagesInverse Kinematics Algorithms for Protein Structure Determinationulysse_d_ithaqu7083No ratings yet
- Plasma Physics Chapter IntroductionDocument18 pagesPlasma Physics Chapter IntroductionNeena JanNo ratings yet
- Molcas TutorialDocument29 pagesMolcas TutorialAnia Beatriz RodriguezNo ratings yet
- Quaccs Scientific Python Module HandoutDocument6 pagesQuaccs Scientific Python Module HandoutDeborah CrittendenNo ratings yet
- HWS Computational PDFDocument34 pagesHWS Computational PDFBheim LlonaNo ratings yet
- ComputationalAstrophysicsL6 PDFDocument20 pagesComputationalAstrophysicsL6 PDFAlirezakhNo ratings yet
- MIT8 04S13 ps9Document5 pagesMIT8 04S13 ps9Yul LopeNo ratings yet
- Jnu TalkDocument35 pagesJnu Talkapi-242448077No ratings yet
- Lecture Notes For: Solid State Electronic Devices: With Modifications by M. Nadeem Akram and Per OhlckersDocument45 pagesLecture Notes For: Solid State Electronic Devices: With Modifications by M. Nadeem Akram and Per OhlckersSaleha QuadsiaNo ratings yet
- Bonding in Molecules 2019 2nd Yr Michaelmas TermDocument71 pagesBonding in Molecules 2019 2nd Yr Michaelmas Termtresa.george27No ratings yet
- Gaussian Basis SetsDocument40 pagesGaussian Basis SetsIsmael Antonio Gonzalez RamirezNo ratings yet
- Summary of Quantum Field Theory: 1 What Is QFT?Document4 pagesSummary of Quantum Field Theory: 1 What Is QFT?shivsagardharamNo ratings yet
- Handout on Tight-binding Calculation for Band StructuresDocument5 pagesHandout on Tight-binding Calculation for Band StructuresalkeroneNo ratings yet
- Core Course: What You Should KnowDocument4 pagesCore Course: What You Should KnowjalajsinghNo ratings yet
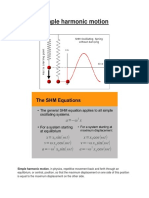
- Simple Harmonic MotionDocument13 pagesSimple Harmonic MotionKifaya Khan الكفايةNo ratings yet
- A.J.Leggett - The Fractional Quantum Hall Effect: Laughlin Wave Function, Fractional Charge and StatisticsDocument11 pagesA.J.Leggett - The Fractional Quantum Hall Effect: Laughlin Wave Function, Fractional Charge and StatisticsGreamxxNo ratings yet
- Computational Physics - Exercise 5: 0 Python PrimerDocument2 pagesComputational Physics - Exercise 5: 0 Python PrimerToon PillaertNo ratings yet
- Doping induced effect on optical and band structure properties of SrLiAl3N4Eu+2 based PhosphorsDocument32 pagesDoping induced effect on optical and band structure properties of SrLiAl3N4Eu+2 based PhosphorsSikander AzamNo ratings yet
- DFT Software: Who, What, and How MuchDocument5 pagesDFT Software: Who, What, and How MuchFaheem RajuNo ratings yet
- Firefly 8.2.0 Keyword List: Firefly - Input - Rev002 PDFDocument192 pagesFirefly 8.2.0 Keyword List: Firefly - Input - Rev002 PDFSiddheshwar Chopra100% (1)
- JC Cuevas DFTDocument59 pagesJC Cuevas DFTMaría Díazgranados JiménezNo ratings yet
- Basis Set Excel LectureDocument11 pagesBasis Set Excel LectureisohsoNo ratings yet
- Bulk Modulus Paper PDFDocument307 pagesBulk Modulus Paper PDFAlbertNo ratings yet
- The Chemistry of Organic Germanium Tin and Lead Compounds Vol-1-1995-PataiDocument1,000 pagesThe Chemistry of Organic Germanium Tin and Lead Compounds Vol-1-1995-Pataieburbano12No ratings yet
- Orca Manual 4 1 0 PDFDocument996 pagesOrca Manual 4 1 0 PDFSiddheshwar ChopraNo ratings yet
- Gaussian TutorialDocument36 pagesGaussian TutorialandreaNo ratings yet
- Homo LumoDocument12 pagesHomo LumoShivam KansaraNo ratings yet
- Density Functional TheoryDocument21 pagesDensity Functional TheoryDian KurniawatiNo ratings yet
- PhysRevB 50 17953Document27 pagesPhysRevB 50 17953Paramita HaldarNo ratings yet
- Infrared, Thermochemistry, UV-Vis, and NMR MethodsDocument3 pagesInfrared, Thermochemistry, UV-Vis, and NMR Methodsfateixeira77No ratings yet
- Predicting The Properties of Drug Molecules:: Quantum Mechanics and Molecular MechanicsDocument45 pagesPredicting The Properties of Drug Molecules:: Quantum Mechanics and Molecular MechanicsekaipNo ratings yet
- Lobster Users Guide 1.2.0Document28 pagesLobster Users Guide 1.2.0Minhaj GhouriNo ratings yet
- OrcaManual 2.7.0Document418 pagesOrcaManual 2.7.0Kristhian Alcantar MedinaNo ratings yet
- Tin (IV) Compounds of Tridentate Thiosemicarbazone Schiff Bases Synthesis, Characterization, In-Silico Analysis and in Vitro CytotoxicityDocument12 pagesTin (IV) Compounds of Tridentate Thiosemicarbazone Schiff Bases Synthesis, Characterization, In-Silico Analysis and in Vitro CytotoxicityGustavo MartinsNo ratings yet
- DMol3 Density-Functional Standard ToolDocument32 pagesDMol3 Density-Functional Standard ToolJosé Luis Rosas Huerta100% (1)
- 2022 Question Paper MMDDDocument5 pages2022 Question Paper MMDDKD GaMiNgNo ratings yet
- QMMM Tutorial Ws10 11Document36 pagesQMMM Tutorial Ws10 11weilaiNo ratings yet
- WIEN2k Code 2 PDFDocument282 pagesWIEN2k Code 2 PDFJaqen H'ghar100% (1)
- Paper Isomerization Nitrito Complejos CoDocument7 pagesPaper Isomerization Nitrito Complejos CoJuan Gabriel FernándezNo ratings yet
- Chemkin PDFDocument44 pagesChemkin PDFmohamedIGCMONo ratings yet
- Gabedit-A Graphical User Interface For Computational Chemistry SoftwaresDocument9 pagesGabedit-A Graphical User Interface For Computational Chemistry SoftwaresDenesis TejedaNo ratings yet
- Zmat TutorialDocument27 pagesZmat TutorialДьего Фернандо0% (1)
- Acs JCTC 7b00118Document21 pagesAcs JCTC 7b00118chrisselrNo ratings yet
- Jurnal Amoeba (Inggris)Document16 pagesJurnal Amoeba (Inggris)Taufiq AliNo ratings yet
- Crystal09 ManualDocument306 pagesCrystal09 ManualDaveyNo ratings yet
- Best Practice DFT Protocols For Basic Molecular Computational ChemistryDocument23 pagesBest Practice DFT Protocols For Basic Molecular Computational ChemistryPedro HenriqueNo ratings yet