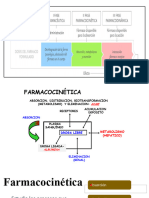
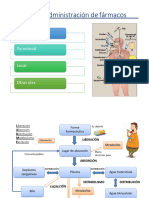
You might also like
- DocxDocument5 pagesDocxMichael ValdezNo ratings yet
- QF - Yuri Vladimir Carrasco ReateguiDocument47 pagesQF - Yuri Vladimir Carrasco ReateguiYuvil ReateguiNo ratings yet
- Farmacocinética: el estudio del recorrido de los fármacosDocument32 pagesFarmacocinética: el estudio del recorrido de los fármacosMichael Jefri Villar100% (1)
- AnticonvulsivantesDocument21 pagesAnticonvulsivantesAlejandraRodriguezNo ratings yet
- Farmacología de Las Hormonas Tiroideas CuadrosDocument10 pagesFarmacología de Las Hormonas Tiroideas CuadrosKevin Quispe OchoaNo ratings yet
- PRACTICA 2 LaboratorioDocument5 pagesPRACTICA 2 LaboratoriocarlosNo ratings yet
- Fármacos Acción EspecíficaDocument32 pagesFármacos Acción EspecíficaXadvier Rivera Jimenez100% (2)
- Farmaco 1 BiotransformacionDocument135 pagesFarmaco 1 BiotransformacionSamyr Domingos50% (2)
- Evaluación Sobre FarmacocinéticaDocument5 pagesEvaluación Sobre FarmacocinéticaNieves VargasNo ratings yet
- Características Generales de Los PlaguicidasDocument17 pagesCaracterísticas Generales de Los Plaguicidassuroma100% (1)
- Alimentos funcionales, más que nutriciónDocument27 pagesAlimentos funcionales, más que nutriciónLeonel EstradaNo ratings yet
- Acciones de Los Fármacos en El OrganismoDocument10 pagesAcciones de Los Fármacos en El OrganismoRuBy RomeroNo ratings yet
- Manual LMCCMDocument80 pagesManual LMCCMcirce hwang0% (1)
- Tema 12. Farmacologà A Del SNCDocument16 pagesTema 12. Farmacologà A Del SNCAdrianaNo ratings yet
- Farmacocinetica y FarmacodinamiaDocument49 pagesFarmacocinetica y FarmacodinamiaHenry Anival Callampi VilcaNo ratings yet
- Farmacia Hospitalaria Sistema de Dispensación de Medicamentos en Dosis Unitarias (Sdmdu) - Semana 04 Part 1Document27 pagesFarmacia Hospitalaria Sistema de Dispensación de Medicamentos en Dosis Unitarias (Sdmdu) - Semana 04 Part 1Richard Palomino LujanNo ratings yet
- Estimulantes Del SNC ESTE ESDocument29 pagesEstimulantes Del SNC ESTE ESleonard_cantillo_1No ratings yet
- Formas FarmacéuticasDocument21 pagesFormas FarmacéuticasmarthaescobarNo ratings yet
- FarmacodinamiaDocument7 pagesFarmacodinamiaNachoo PorteelaNo ratings yet
- Farmacos CanalesDocument42 pagesFarmacos CanalesVictor OntonNo ratings yet
- Importancia farmacovigilancia pediatríaDocument15 pagesImportancia farmacovigilancia pediatríaJos VarNo ratings yet
- AntifúngicosDocument5 pagesAntifúngicosAgostina Velia DamianiNo ratings yet
- Semana - 5Document38 pagesSemana - 5Elva Roxana Chavez LiñanNo ratings yet
- Farmacos OftalmologicosDocument5 pagesFarmacos OftalmologicosVictoria JijonNo ratings yet
- AntiepilépticosDocument83 pagesAntiepilépticosCátedra de Farmacología de la Escuela de Medicina de La UNIVERSIDAD DEL ZULIANo ratings yet
- Formas Farmaceúticas - SólidosDocument36 pagesFormas Farmaceúticas - SólidosNatalia BahamonNo ratings yet
- Proteínas y péptidos bioactivosDocument54 pagesProteínas y péptidos bioactivosFelipe Martínez ArbeláezNo ratings yet
- Preparaciones Extractivas de Productos Naturales y Extractos Estandarizados.Document2 pagesPreparaciones Extractivas de Productos Naturales y Extractos Estandarizados.jessicaNo ratings yet
- Resumen Sólidos OralesDocument7 pagesResumen Sólidos OralesIvan MonroyNo ratings yet
- RECUBRIMIENTO EntéricoDocument3 pagesRECUBRIMIENTO EntéricoRUBYNo ratings yet
- El circuito de recompensa y la dopaminaDocument4 pagesEl circuito de recompensa y la dopaminaLourdes Alejandra Velazquez VeraNo ratings yet
- FARMACOGNOSIADocument13 pagesFARMACOGNOSIAMargotPilarHuamanìHinostrozaNo ratings yet
- Ansiolíticos No BenzodiacepínicosDocument5 pagesAnsiolíticos No BenzodiacepínicosJunior ForttiniNo ratings yet
- Mecanismos de Absorcion y Transporte de FarmacosDocument8 pagesMecanismos de Absorcion y Transporte de FarmacosMiguel AngelNo ratings yet
- Medicamentos Antineoplasicos Cancer Ii ParteDocument5 pagesMedicamentos Antineoplasicos Cancer Ii PartePedro José Ortíz OrtegaNo ratings yet
- Gestión MedicamentosDocument74 pagesGestión Medicamentosjuan migel herrrera fernandezNo ratings yet
- Intox - ParacetamolDocument31 pagesIntox - ParacetamolJhon GomezNo ratings yet
- Preparaciones parenterales: concepto, clasificación, ventajas, desventajas y requisitos para su elaboraciónDocument16 pagesPreparaciones parenterales: concepto, clasificación, ventajas, desventajas y requisitos para su elaboraciónMarvin A. GonzalezNo ratings yet
- Aines No SelectivoDocument18 pagesAines No SelectivoJulio RamirezNo ratings yet
- SOLUBILIDAD y CINÉTICADocument76 pagesSOLUBILIDAD y CINÉTICAAnaNo ratings yet
- 1257745432.power Farmaco - Iecas.1Document44 pages1257745432.power Farmaco - Iecas.1AdriianaLaraNo ratings yet
- LIDOCAINADocument14 pagesLIDOCAINAJanet pilar Gonzales ramirezNo ratings yet
- Consideraciones farmacológicas en pacientes con enfermedad renal crónica (ERCDocument13 pagesConsideraciones farmacológicas en pacientes con enfermedad renal crónica (ERCLucia Alvarez GarciaNo ratings yet
- Fármacos Del Sistema Nervioso CentralDocument7 pagesFármacos Del Sistema Nervioso CentralMarcela BermudezNo ratings yet
- Elaboracion de Formas SolidasDocument29 pagesElaboracion de Formas SolidasPedro Pablo TorresNo ratings yet
- Analgésicos Opioides. Morfina. Agonistas y AntagonistasDocument85 pagesAnalgésicos Opioides. Morfina. Agonistas y AntagonistasJazitaRomeroNo ratings yet
- CumarinicosDocument67 pagesCumarinicosJaime RevillaNo ratings yet
- RelaciónDosisEfectoDocument17 pagesRelaciónDosisEfectoSofia Guadalupe Lerma CarreónNo ratings yet
- Sistemas dispersos: soluciones, coloides y suspensionesDocument7 pagesSistemas dispersos: soluciones, coloides y suspensionespeter_esscribdNo ratings yet
- Asignación en Equipo PDFDocument8 pagesAsignación en Equipo PDFBrian PiundoNo ratings yet
- Medicamentos y Técnicas de AplicaciónDocument59 pagesMedicamentos y Técnicas de Aplicaciónerazobritney181No ratings yet
- Estructura de los glúcidos: monosacáridos, ciclos y epímerosDocument14 pagesEstructura de los glúcidos: monosacáridos, ciclos y epímerosDARIOHIDALGONo ratings yet
- Clasificacion ATCDocument2 pagesClasificacion ATCmiguel moralesNo ratings yet
- Farmacología 1 GeneralidadesDocument26 pagesFarmacología 1 GeneralidadesKARLA NICOL FIGUEROA ARIASNo ratings yet
- Caracteristicas de La Calidad de Los MedicamentosDocument26 pagesCaracteristicas de La Calidad de Los MedicamentosKatherine OjedaNo ratings yet
- Farmacologia Semana 2 UnwDocument34 pagesFarmacologia Semana 2 UnwJade Nicel Liliana Esquivel MogollonNo ratings yet
- Farmacocinética 2023 IiDocument35 pagesFarmacocinética 2023 IiAngie Acosta RodriguezNo ratings yet
- Clase 2 Farmacologia 2S 2020Document29 pagesClase 2 Farmacologia 2S 2020Cha GamaNo ratings yet
- Biología Molecular y Celular: Una guía introductoria para aprender Biología Celular y MolecularFrom EverandBiología Molecular y Celular: Una guía introductoria para aprender Biología Celular y MolecularNo ratings yet
- Rejuvenecer Con El Plasma Sanguíneo De Los JóvenesFrom EverandRejuvenecer Con El Plasma Sanguíneo De Los JóvenesRating: 5 out of 5 stars5/5 (1)
- Medicina LegalDocument18 pagesMedicina LegalEberth VasquezNo ratings yet
- PDF 20220406 143601 0000Document40 pagesPDF 20220406 143601 0000Alexandra PedrazaNo ratings yet
- Regulación Génica AbejasDocument4 pagesRegulación Génica AbejasEnzo Claudio Leandro Soto Rojas100% (1)
- Medio MIODocument10 pagesMedio MIOJose Maria RojasNo ratings yet
- Consentimiento InformadoDocument3 pagesConsentimiento Informadoluarduse11081987No ratings yet
- Breve historia de la microbiologíaDocument9 pagesBreve historia de la microbiologíaPaula DanielaNo ratings yet
- HerenciaDocument68 pagesHerenciaCande AntónNo ratings yet
- OrtotoluidinaDocument6 pagesOrtotoluidinaRodrigo GomezNo ratings yet
- Guia Del Cuidado de Polinizadores de ArándanoDocument9 pagesGuia Del Cuidado de Polinizadores de ArándanomarcosanchezveramendNo ratings yet
- 21Document3 pages21Fabián Bermúdez ValdezNo ratings yet
- Sistema Oseo - Leila RiosDocument14 pagesSistema Oseo - Leila RiosLeila RiosNo ratings yet
- SEMINARIO UltimoDocument6 pagesSEMINARIO UltimoMaríaAntezana0% (1)
- NeoplasiasDocument5 pagesNeoplasiasjose colon machucaNo ratings yet
- RIo MissisipiDocument6 pagesRIo Missisipicristina_ropero6No ratings yet
- Actividad de Aprendizaje #13Document4 pagesActividad de Aprendizaje #13Evaristo Bernilla CajoNo ratings yet
- Dis LaliaDocument50 pagesDis LaliaHenri Gino Vera PlasenciaNo ratings yet
- Comunicación CelularDocument17 pagesComunicación CelularOscar Ivan Salas DelgadoNo ratings yet
- Pistas PlanasDocument6 pagesPistas PlanasOscar RodríguezNo ratings yet
- Crucigrama VeterianariaDocument2 pagesCrucigrama VeterianariaJenny HernandezNo ratings yet
- Importancia de La SangreDocument2 pagesImportancia de La SangreJEANELA BRAVO RAMOSNo ratings yet
- Composición Química Del Aceite Esencial de Hojas de Oregano (Origanum Vulgare)Document6 pagesComposición Química Del Aceite Esencial de Hojas de Oregano (Origanum Vulgare)Cesar MartinezNo ratings yet
- Actividad 3 Higiene IndustrialDocument3 pagesActividad 3 Higiene IndustrialALLISON DANIELA NIEVES MEJIANo ratings yet
- Rheinberger - 2015 - Partículas Citoplásmicas. Trayectoria de Un Objeto Científico PDFDocument18 pagesRheinberger - 2015 - Partículas Citoplásmicas. Trayectoria de Un Objeto Científico PDFIesus GudiñoNo ratings yet
- Guia de Setas de Salamanca II (Tribuna - Enusa)Document18 pagesGuia de Setas de Salamanca II (Tribuna - Enusa)Fernando RodriguezNo ratings yet
- Grupo N°1Document35 pagesGrupo N°1Toñito EcNo ratings yet
- Olericultura GeneralDocument11 pagesOlericultura GeneralDepartamento de Fitotecnia100% (1)
- Labo 3 Micro IIDocument40 pagesLabo 3 Micro IIMargaritaNo ratings yet
- Modelos y Teorias de Enfermeria ComunitariaDocument17 pagesModelos y Teorias de Enfermeria ComunitariaHospital Oncologico92% (25)
- Tasas de PrevalenciaDocument9 pagesTasas de PrevalenciaRonald PensamientoNo ratings yet
- 6 Práctica de Ciencias Naturales de 4to. Del BachilleratoDocument3 pages6 Práctica de Ciencias Naturales de 4to. Del BachilleratoSteven RodríguezNo ratings yet